事業評価書(中間)
| 事務事業名 | 医薬品医療用具審査システム | |||||||||||||||||||||||||||||||||||||||||
| 事務事業の概要 | (1)目的 | 品質、有効性及び安全性の高い新医薬品・医療用具を迅速に承認する。 | ||||||||||||||||||||||||||||||||||||||||
| (2)内容 |
承認申請が行われた医薬品及び医療用具の品質、有効性及び安全性について、倫理性、科学性及び信頼性の確保された申請資料を、その時点における医学・薬学等の見地から、一定の審査期間内に評価し、品目毎に承認を与えるもの。 (1) 治験の円滑な推進 医薬品開発の最終段階で実施されるヒトを対象とした臨床試験(治験)の実施に当たり、被験者の人権とデータの信頼性等について定められた基準(GCP省令)を満たすよう指導を行う。また、事前に治験相談を行うことにより、承認審査の観点から必要な資料の作成を促進する。 (2) 医薬品等の承認審査 医薬品等の承認審査に当たっては、以下に重点を置いている。
(3) 医薬品等の再審査 承認時までの限られた成績に基づき承認した内容が、医療実態、小児、及び高齢者等において適切であるか否かを確認し、必要があれば承認内容変更等の措置を行う。 (4) 医薬品等の再評価 医学・薬学の学問的水準の進歩に応じ、既承認医薬品の品質、有効性及び安全性の見直しを行い、必要があれば承認内容の変更及び承認取消等の措置を行う。
|
|||||||||||||||||||||||||||||||||||||||||
| (3)達成目標 |
(1) 医療上特に必要性が高い新医薬品・医療用具の優先審査を進め、諸外国で既に承認され、その有効性及び安全性が高く評価されている品目等を通常の品目より早く承認する。 (2) 承認申請された医薬品等について、定められた標準事務処理期間内に処理する。
(3) リスクの高いものを重点的に審査するため、リスクの低い医療用具については基準を定めて自己認証制度等の対象とし、承認審査の効率化を図る。 (4) 再審査期間中に収集された成績に基づき、必要に応じて承認内容を適切に改める。 (5) 適切な再評価の実施により、品質、有効性及び安全性の観点から不適当な医薬品について、承認内容の変更及び承認取消等の措置を行う。 |
|||||||||||||||||||||||||||||||||||||||||
| 評価 | (1)必要性 |
〔国民や社会のニーズに照らした妥当性〕 適切な治験の実施は、医薬品等の有効性及び安全性に関する正確なデータ収集の前提であり、国民にとって有益な医薬品等が市場に供給されることに資するとともに、被験者として治験に参加する患者の安全の保護に資する。 ○医薬品等の承認審査 医薬品等の品質、有効性及び安全性を十分に評価しつつ、迅速に承認を行うことは、適切な国民医療の実施に不可欠。 <医薬品等分類別申請件数と処理件数(平成11年実績)>
<外国との承認審査に係る人員比較(2000年)>
なお、迅速化・効率化を図る一方で、特に安全性等については、様々なヒト又は動物由来医薬品等の登場により、承認審査の段階における知見では評価困難なものも増えており、既存の枠組みにとらわれず、市販後対策を強化することを含め、現行の製造承認制度の見直しをも視野に入れた対策を検討することが必要。 ○再審査・再評価 新医薬品等については、小児及び高齢者等に係る情報が承認時までには極めて少ないことから、承認後の情報に基づき、さらなる適正使用確保措置が講じられるよう、承認内容を見直すことが必要。 〔公益性〕 治験は、将来の国民(患者)に広く用いられる医薬品等の有効性及び安全性を確認・検証するために行われるものであり、また、被験者として治験に参加する患者は、将来の国民(患者)のために自発的に協力していることから、高い公益性を有する仕組みとして、医薬品の承認審査に不可欠。 ○医薬品等の承認審査 国民医療全体において広く使用される医薬品等の品質、有効性及び安全性を審査し、迅速に承認することは、国民医療の適切な実施に不可欠。 ○再審査・再評価 医療において不可欠な薬物療法等を行うに際し、その時点の最新の知見に照らし、品質、有効性及び安全性が確認されている薬物により治療を受けられるよう環境整備を図ることは極めて公益性が高い。 〔官民の役割分担〕 治験は、製薬企業、医療機関等が主体となって実施されるものであり、他方、適正な治験が効率的に実施されるよう薬事法等の規制に基づいて指導することは行政の役割。 ○医薬品等の承認審査 医薬品等の品質、有効性及び安全性に関する情報収集及びそれに基づく承認申請は、製薬企業により行われるものであり、他方、申請資料の信頼性の確認及び客観的評価を公平な立場から行うことは、行政の役割。 ○再審査・再評価 医薬品等の品質、有効性及び安全性については承認を得ている企業においてこれを証明する資料の収集・評価を行うものであり、他方、これらの信頼性の確認及び客観的評価を公平な立場で行うことは、行政の役割。 〔国と地方の役割分担〕 医薬品の承認審査等には高度の専門性が必要であることから、国が行うべき。また、通常、治験は全国的に実施され、医薬品等は全国的に流通、使用されることから、これらの指導、評価等は国が一元的に行うべき。なお、かぜ薬等一部の医薬品については、既に基準を定めて都道府県知事承認としており、適切な役割分担が確保されている。 〔民営化や外部委託の可否〕 提出される資料には企業秘密に属するものが多数含まれること、客観的かつ最新の学問的知見から公平な評価を行う必要があることから、国が行うべき。 〔緊要性の有無〕 品質、有効性及び安全性を審査し、医薬品等を迅速に承認するためのものであり、緊要性がある。 ○再審査・再評価 医薬品等に係る安全対策の一環であることから緊要性のあるものである。 〔他の類似施策(他省庁分も含む)〕 〔社会経済情勢の変化を受けた廃止、休止の可否(継続事業のみ)〕 医薬品等の承認及び使用は医療に必要不可欠なものとして間断なく行われるものであり、制度全体について廃止、休止を行うことは不可能。 |
||||||||||||||||||||||||||||||||||||||||
| (2)有効性 |
〔これまで達成された効果(継続事業)、今後見込まれる効果〕 平成9年に施行された新GCPを遵守した治験が実施されることにより、質の高い有効性及び安全性に係るデータが収集されることが期待されるとともに、治験中の被験者(患者)の安全がより高いレベルで守られる。 ○医薬品等の承認審査 標準事務処理期間を18か月から欧米並みの12か月に短縮した。 ○再審査・再評価 治験段階でのデータは患者数、年齢等の点で限られたものとなるので、承認後さらに十分なデータが追加された段階で、最新の知見をもって、医薬品の品質、有効性及び安全性を見直すことは有効。 <再審査の結果> これまでに、1,940品目について再審査を行い、139品目を承認事項変更としている。 <再評価結果>
新再評価のうち、品質再評価事業については、99成分1,288品目について終了しており、149品目承認整理を行っている。 〔効果の発現が見込まれる時期〕 |
|||||||||||||||||||||||||||||||||||||||||
| (3)効率性 |
〔単年度の費用〕 医薬品等の承認審査事務においては、品質、有効性及び安全性の確保に十分留意して厳格な審査を不断に行っており、審査業務の一層の効率化、透明化の要請に応えるとともに、承認審査事務の迅速化、効率化及び高度化を図るべく、恒常的にシステムを運営することが必要であって、単年度費用に基づく検討になじまない。 〔手段の適正性〕 治験の段階で事前相談を受け付け、審査の観点から必要なデータを収集させるなど効率化を図っている。 <優先審査承認品目の割合(平成12年実績)> 平成12年に承認された新医薬品に占める優先審査対象品目の割合 <標準事務処理期間内処理品目数(平成11年に事務処理を終了したもの)>
(注:上記品目は、電話による照会又はファックスによる照会に係る期間を除かずに集計したものであり、これらの期間を適切に考慮すれば、該当する品目は増大する。また、標準事務処理期間の期間計測方法については検討中。) <都道府県への権限委譲又は自己認証を行うために必要な基準作成件数> これまで医薬品については15基準を制定し都道府県知事承認に、医療用具については100基準を制定し承認不要にしており、リスクの高いものにより重点を置いた審査を行うことにより、審査の効率化・迅速化を図っている。 ○再審査・再評価 治験段階でのデータは患者数、年齢等の点で限られたものとならざるを得ないが、一方で有効な医薬品を迅速に承認することは社会的な要請である。このような中、一旦承認した医薬品を承認後十分なデータが追加された段階で最新の知見をもって見直すことは効率的。 |
|||||||||||||||||||||||||||||||||||||||||
| (4)その他 (公平性・優先性など) |
○薬事法の規定に基づき、医薬品等の承認・許可を申請する事業者は、申請に対する審査に要する実費の額を考慮した額(手数料)を納めることとされている。なお、平成13年度の手数料収入は約618百万円を見込んでいる。 <手数料の分類> 1.許可手数料 2.承認手数料 3.一部変更承認手数料 4.再審査手数料 5.業許可証書換・再交付手数料 |
|||||||||||||||||||||||||||||||||||||||||
| 関連事務事業 | なし | |||||||||||||||||||||||||||||||||||||||||
| 特記事項 | 〔各種政府決定との関係及び遵守状況〕 1 経済構造の改革と創造のためのプログラム(平成8年12月閣議決定)において、平成9年度以降3年間にすべての規格の必要性等について検討を行うことが決定されたことから、JIS規格について、ゼロベースでの見直しを行っている。 2 規制改革推進3か年計画(平成13年3月閣議決定)において、基準認証等について、自己確認及び第三者認証を基本とした制度への移行を推進することとされている。 3 規制緩和推進3か年計画(再改訂:平成12年3月閣議決定)について、措置状況が公表(平成13年4月)されている。 〔予算の執行状況(不用、繰越)〕 本事業事務にかかる平成12年度不用額等 (項)厚生本省 (目)医薬品審査等業務庁費 194,998千円 平成12年度システム開発として予算化された「医薬品輸入確認システム」経費については、医薬品業許可台帳の整備とデータの一元化を図る必要があるが、これらの整備には数年間を要することが判明したため、本システムを開発したとしても当分の間運用ができないことから、見合わせたものである。 |
|||||||||||||||||||||||||||||||||||||||||
| 主管課 及び関係課 |
(主管課)医薬局 審査管理課 | |||||||||||||||||||||||||||||||||||||||||
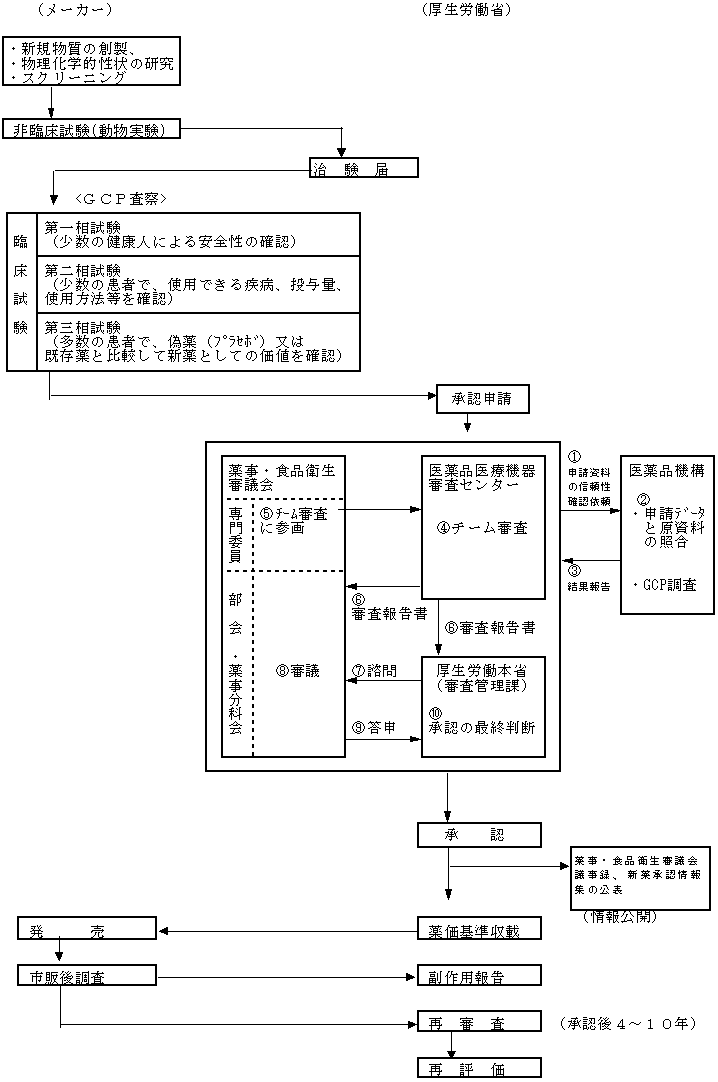
新医薬品・新医療用具の承認審査の流れ
ア.新薬の開発、承認審査、市販後調査等の流れ
*GCP査察は承認申請後に実施。
イ.新医療用具の開発、承認審査、市販後調査等の流れ
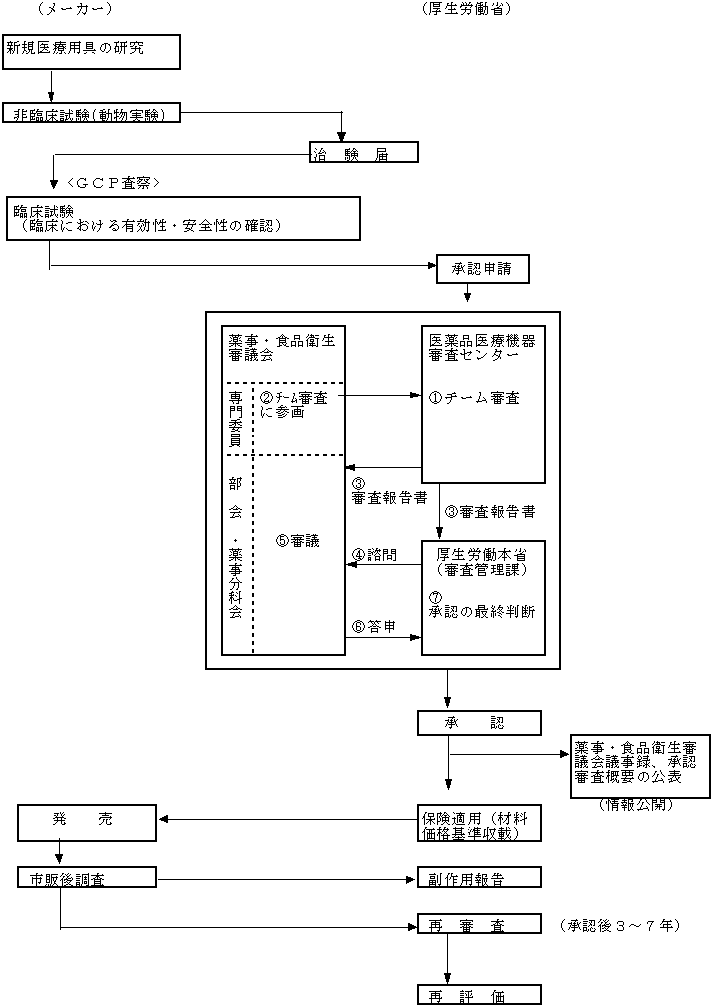
*GCP査察は承認申請後に実施。