ホーム > 政策について > 分野別の政策一覧 > 健康・医療 > 食品 > 食品添加物 > 食品添加物の指定及び使用基準改正に関する指針について > 食品添加物の指定及び使用基準改正に関する指針(別添)
食品添加物の指定及び使用基準改正に関する指針(別添)
※本指針は「「食品添加物の指定及び使用基準改正に関する指針」の一部改正について」(令和4年9月29日付け生食発0929第3号)により改正されています。こちら [267KB]に掲載している改正後の指針をご覧ください。
(別添)
食品添加物の指定及び使用基準改正に関する指針
I 目的
本指針は、食品衛生法第6条により食品添加物として用いることを目的とする化学的合成品を定める場合並びに同法第7条第1項により食品添加物の使用の方法につき基準を定める場合の要請手続、要請書に添付すべき安全性に関する試験成績等必要な資料の範囲、及び資料を作成するために必要な試験の標準的な実施方法等を規定するものである。
II 食品添加物の指定及び使用基準改正に関する基本的考え方
食品添加物は、人の健康を損なうおそれがなくかつその使用が消費者に何らかの利点を与えるものでなければならない。
従って、食品添加物の指定及び使用基準改正に当たっては、次の点が科学的に評価されることが必要である。このため、FA0/WHO合同食品規格委員会の基準等を参考にするとともに、わが国の食品摂取の状況等を勘案し、公衆衛生の観点から、科学的評価を食品衛生調査会において行う。
| 1. | 安全性
食品添加物の安全性が、要請された使用方法において、実証又は確認されること。
|
| 2. | 有効性
食品添加物の使用が、次のいずれかに該当することが実証又は確認されること。なお、対象となる食品の製造又は加工の方法の改善・変更が比較的安価に実行可能であり、改善・変更した結果その添加物を使用しないですむ場合を除く。
| (1) | 食品の栄養価を保持するもの。
ただし、(2)に該当する場合又はその食品が通常の食事の中で重要なものでない場合には、食品中の栄養価を意図的に低下させることも、正当と認められる場合がある。 |
| (2) | 特定の食事を必要とする消費者のための食品の製造に必要な原料又は成分を供給するもの。
ただし、疾病の治療その他医療効果を目的とする場合を除く。 |
| (3) | 食品の品質を保持し若しくは安定性を向上するもの又は味覚、視覚等の感覚刺激特性を改善するもの。
ただし、その食品の特性、本質又は品質を変化させ、消費者を欺瞞するおそれがある場合を除く。 |
| (4) | 食品の製造、加工、調理、処理、包装、運搬又は貯蔵過程で補助的役割を果たすもの。
ただし、劣悪な原料又は上記のいずれかの過程における好ましからざる手段若しくは技術(非衛生的なものを含む。)の使用による影響を隠ぺいする目的で使用される場合を除く。 |
|
III 食品添加物の指定又は使用基準改正に係る手続
| 1. | 要請
食品添加物の指定又は使用基準改正を要請する者は、厚生大臣あて、それぞれ別紙様式1又は別紙様式2により要請書を提出することができる。要請書には、当該食品添加物の成分規格案及び使用基準案並びに安全性に関する資料等を添付しなければならない。
なお、要請者が外国に在住する場合には、日本国内において当該要請に関する事項について責任をもって対応できる者(国内連絡先)を明記すること。また、要請書は、直接、厚生省生活衛生局食品化学課に提出すること。
|
| 2. | 成分規格案及び使用基準案の添付
| (1) | 食品添加物の指定を要請する場合には、原則として、成分規格案を要請書に添付する。また、使用基準案は当該食品添加物の使用対象食品、使用量及び使用方法等を限定する必要がある場合に添付する。 |
| (2) | 使用基準の改正を要請する場合には、当該食品添加物の使用基準と要請する使用基準改正案の対照表を要請書に添付する。 |
|
| 3. | 審査
食品添加物の指定又は使用基準改正の要請については、厚生省生活衛生局食品化学課において事務局審査を行い、食品衛生調査会の意見を聴くことが適切であると認められる場合には、当該要請について食品衛生調査会への諮問に必要な事務を開始する。
食品衛生調査会は審査終了後、諮問された事項に関し厚生大臣あて答申を行う。厚生省は、食品衛生調査会の答申を踏まえ、食品衛生法施行規則改正等必要な事務手続を行う。(下記の図参照) なお、食品衛生調査会における審査の過程等において、必要とされる場合には、要請者に資料の追加提出等を求めることがある。
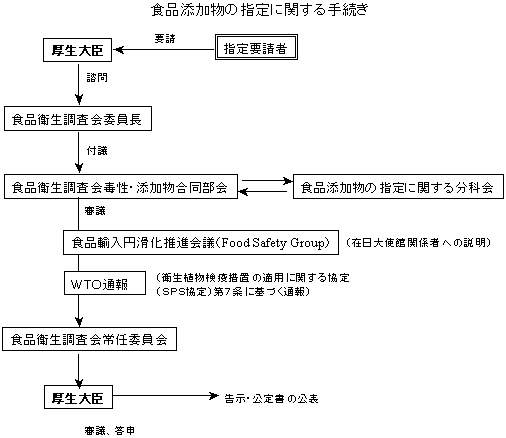
| 注: | 平成15年度の内閣府食品安全委員会の設置により食品添加物の指定等に関する手続は、次のように変更しております。 |
|
| 4. | 標準的事務処理期間
要請書が受理された日から、食品添加物の指定又は使用基準改正までに要する標準的事務処理期間は1年とする。ただし、本期間には、提出された書類又は資料等に不備があり、これを要請者が修正するのに要する期間及び食品衛生調査会等における指摘事項に対し要請者が回答するまでの期間は含まない。 |
IV 食品添加物の指定及び使用基準改正の要請書に添付すべき資料
| 1. | 添付資料の範囲
| (1) | 食品添加物の指定の要請に際しては、原則として、表1に示された資料を添付する。
ただし、当該食品添加物が食品常在成分であるか又は食品内若しくは消化管内で分解して食品常在成分になることが科学的に明らかである場合には、原則として、表1のうち毒性に関する資料の添付を省略することができるが、げっ歯類の28日間反復投与毒性試験及び変異原性試験は添付することが望ましい。
なお、上記ただし書に該当するか否かは、表2の事項について検討の上判断することが必要である。 |
| (2) | 使用基準改正の要請に際しては、原則として、表1において○印を付した資料を添付する。ただし、安全性に関する資料等において△印を付した資料は、食品添加物の指定後に新たに知見が得られた場合及びその他必要な場合に添付すること。 |
| (3) | (1)、(2)にかかわらず、既に指定されている食品添加物と塩基部分のみが異なる又はその異性体である場合その他合理的な理由がある場合は、その理由を説明した上で、適宜資料の添付を省略することができる。 |
| (4) | 要請に係る食品添加物がその要請に係る品質、安全性又は有効性を有することを疑わせる資料については、当該資料の信頼性等にかかわらず、提出しなければならない。 |
|
| 2. | 添付資料作成上の一般的注意
| (1) | 添付資料は、要請者がその責任において提出するものであり、資料内容の信頼性は要請者が確保しなくてはならない。 |
| (2) | 資料概要は邦文で記載されていなければならない。ただし、資料概要以外の添付資料(表1の区分2〜6の資料)については英文で記載されたものであっても差し支えない。 |
| (3) | 添付資料を作成するために必要とされる試験は、試験成績の信頼性を確保するために必要な施設、機器、職員等を有し、かつ適正に運営管理されていると認められる試験施設において実施されなければならない。 |
|
| 3. | 指定要請添付資料の作成上の留意事項
| (1) | 資料概要
| (1) | 資料区分ごとに簡潔にまとめ、各資料との関連を明らかにするよう資料番号を肩に明記する。なお、通しで頁をつけること。 |
| (2) | 表1に示された資料であって、添付を省略した資料については、その理由を記載する。 |
|
| (2) | 起源又は発見の経緯及び外国における使用状況に関する資料
| (1) | 起源又は発見の経緯
要請された品目がいつ、どの国で開発され、その後どの国で食品添加物として使用されるようになったか等、要請に至る経緯を記載する。 |
| (2) | 外国における使用状況
要請された品目の諸外国における許可状況、具体的な使用食品、使用基準、成分規格等を記載すること。併せて、国際機関における安全性評価、使用基準、成分規格等も記載する。 |
|
| (3) | 物理化学的性質及び成分規格に関する資料
食品添加物公定書の通則及び一般試験法等を参考にして、適切な方法により試験した結果に基づき作成する。
| (1) | 名称
一般名、化学名(IUPAC名に準拠する。)等を記載すること。 |
| (2) | 構造式又は示性式
食品添加物公定書を参考に構造式又は示性式を記載すること。 |
| (3) | 分子式及び分子量
食品添加物公定書の通則に準拠すること。 |
| (4) | 含量規格
含量規格は、製造過程、定量誤差及び安定性等に基づき、安全性と有効性に関して同等とみなせる一定品質を保証するのに必要な値を設定する。
食品添加物としての有効成分の含量を%で示し、有効成分が2種以上存在する場合は、それぞれについて記載する。 |
| (5) | 製造方法
製造方法によっては、不純物の種類又は量が異なる可能性もあるので、製造工程を簡明に記載する。 |
| (6) | 性状
性状は、使用時の識別及び取扱い上必要となる事項について、通例、味、におい、色、形状等を記載する。 |
| (7) | 確認試験
確認試験は、当該物質が目的の食品添加物であるか否かをその特性に基づいて確認するための試験である。従って、食品添加物の化学構造上の特徴に基づいた特異性のある試験である必要がある。
確認試験以外の項目の試験によっても食品添加物の確認が可能な場合には、それらを考慮に入れることができる。例えば、定量法に特異性の高いクロマトグラフ法を採用する場合には、確認試験を簡略化することができ、重複する内容で設定する必要はない。
確認試験を行う方法としては、通例、スペクトル分析に基づく方法及び化学反応による方法が考えられる。なお、化学反応については、化学構造の特徴を確認するのに適切なものがある場合に設定する。 |
| (8) | 示性値
示性値とは、吸光度、旋光度、pH及び融点等の物理的化学的方法により測定される数値をいい、品質を確保するうえで必要な項目を記載する。 |
| (9) | 純度試験
純度試験は、食品添加物中の不純物を試験するために行うもので、定量法とともに食品添加物の純度を規定する試験である。食品添加物中に混在する可能性のある物質(原料、中間体、副生成物、分解生成物、試薬・触媒、重金属・無機塩及び溶媒)のうち必要なものを対象とする。 |
| (10) | 乾燥減量、強熱減量又は水分
乾燥減量試験は、乾燥することによって失われる食品添加物中の水分、結晶水の全部又は一部及び揮発性物質等の量を測定するために行う。強熱減量試験は、強熱することによって、その構成成分の一部又は混在物を失う無機物について行う。水分試験は、食品添加物中に含まれる水分含量を知る目的で行う。 |
| (11) | 強熱残留物(強熱残分)
強熱残留物試験は、通例、有機物中に不純物として含まれる無機物の含量を知るために行うが、場合によっては、有機物中に構成成分として含まれる無機物又は熱時揮発する無機物中に含まれる不純物の量を測定するために行う。 |
| (12) | 定量法
定量法は、有効成分の含量を、物理的、化学的又は生物学的方法により測定する試験である。相対的な試験方法を設定する場合には、定量試験に用いる標準物質について規格を設定する。
正確さ、再現性及び特異性を重視して、試験法を設定する。ただし、特異性の低い方法であっても、適切な純度試験により、混在物の限度が規制されている場合には、再現性のよい絶対量を測定しうる試験方法を設定して差し支えない。その場合には、特異性にかける部分について、純度試験等に特異性の高い方法を用いることにより、相互に補完し合うことが必要である。
なお、定量しようとする成分が2種以上ある場合は、重要なものから記載する。 |
| (13) | 食品添加物の安定性
食品添加物の安定性について、分解物の検索を含め、検討を行う。 |
| (14) | 食品中の食品添加物の分析法
原則として、食品添加物を使用する可能性の高い食品につき、当該食品の化学分析等によりその添加を定性的及び定量的に確認できる方法を設定する。なお、同様の目的をもつ他の食品添加物等との分離定量に留意すること。
ただし、使用基準を設定する必要がない場合又は食品中に残留しない場合にあっては、食品中の食品添加物の分析法のうち、定量法の設定を省略することができる。 |
| (15) | 成分規格案の設定根拠
| ア) | 成分規格案は、国際機関によって設定された成分規格を参考とし、 上記(1)〜(12)の資料に基づき、当該食品添加物の安全性、有効性に関し、一定の品質を担保するために必要なものを設定する。 |
| イ) | 国際機関によって設定された成分規格及び諸外国の成分規格と成分規格案との対照表を添付する。 |
|
|
| (4) | 有効性に関する資料
| (1) | 有効性に関する試験については、食品添加物の用途ごとに期待する効果があることを裏付ける試験を行う。
例えば、酸化防止剤については、対象食品に関する抗酸化効果が添加量及び時間経過との関係において明らかになるような試験を行うべきであり、保存料については、対象食品に対する保存性向上の効果が明らかとなるような試験を行う必要がある。 |
| (2) | 既に指定されている同様の用途の食品添加物がある場合は、それらの食品添加物と効果を比較することが望ましい。 |
| (3) | 食品添加物の食品中における安定性に関する試験を行う。なお、安定でない場合は、主な分解物の種類及び生成程度について検討すること。 |
| (4) | 食品添加物の食品中の主要な栄養成分に及ぼす影響についても検討する。 |
|
| (5) | 安全性に関する資料
| (1) | 毒性に関する資料
| ア) | 毒性試験データの信頼性を確保するため、これらの試験は医薬品の安全性試験の実施に関する基準等、適切なGLP(Good Laboratory Practice)に従って実施されなくてはならない。 |
| イ) | 各々の毒性試験については、食品添加物の安全性の適正な評価に資するよう、標準的な実施方法を第V章に示した。
しかし、本来、すべての食品添加物について一律の試験方法を定めることは合理的でなく、また、今後とも科学技術の進歩に応じ新しい試験方法の開発が行われることも考えられるので、得られた所見が食品添加物の安全性評価に資するものである限り、必ずしも第V章に定めた方法に固執するものではない。
例えば、OECDガイドライン、米国FDAガイドラインに準拠した試験は、食品添加物の安全性評価にとって基本的に問題ないものと考えられる。 |
| ウ) | 90日反復投与毒性試験をげっ歯類1種又は非げっ歯類1種について実施した場合には、それぞれに相当する動物種に係る28日反復投与毒性試験の実施を省略することができる。 |
| エ) | 1年間反復投与毒性試験、発がん性試験を各々所要の動物種について実施した場合には、1年間反復投与毒性/発がん性併合試験を実施する必要はない。
また、1年間反復投与毒性/発がん性併合試験をげっ歯類1種について実施した場合には、1年間反復投与毒性試験及び発がん性試験のげっ歯類1種について試験の実施を省略することができる。 |
| オ) | 食品添加物の分解物及び混在する不純物の安全性についても、必要に応じ、検討を行う。 |
|
| (2) | 体内動態に関する資料
| ア) | ヒトが摂取した場合の生体内における吸収、分布、代謝、排泄を推定するため、体内動態に関する試験を実施する。従って、動物試験結果をまとめるのみでなく、ヒトにおける体内動態や有害な作用の発現の推定等について考察を行わなくてはならない。 |
| イ) | 体内動態に関する試験の標準的な実施方法も、第V章に示したところであるが、その取扱いについては、上記(1) イ)に述べた毒性試験の場合と同様である。 |
|
| (3) | 食品添加物の一日摂取量に関する資料
| ア) | 食品添加物の一日摂取量は、使用対象食品の一日摂取量に食品添加物の使用量を乗じて求める。食品の一日摂取量は、国民栄養調査の食品群別摂取量又はその他の資料等により適切に推定する。 |
| イ) | 当該食品添加物の安全性について、一日摂取量と毒性試験から求められる一日摂取許容量との比較等につき考察する。なお、考察に当たっては、同種の食品添加物等が併せて摂取される場合等の安全性についても検討すること。
また、我が国の食物摂取の実態を踏まえ、栄養成分の過剰摂取や電解質バランスへの影響等についても、必要に応じ、検討すること。 |
|
|
| (6) | 使用基準案に関する資料
| (1) | 食品添加物の安全性、有効性を総合的に検討し、使用対象食品及び使用量等を限定するため、使用基準を設定する必要があると判断した場合には、当該使用基準を設定する根拠を上記(2)〜(5)の資料に基づき明らかにすること。なお、使用基準案は、既に設定されている他の食品添加物の使用基準を参考に作成すること。 |
| (2) | 使用基準を設定する必要がないと判断した場合には、上記(2)〜(5)の資料に基づき、その根拠を明らかにする。 |
|
|
| 4. | 使用基準改正要請添付資料の作成上の留意事項
「3.指定要請書の添付資料の作成における留意事項」に準ずる。ただし、使用基準案の設定に関する資料においては、要請した使用対象食品の追加、使用量の変更等、使用基準を改正する根拠を、資料に基づき明らかにする。 |
| 表1 | 食品添加物の指定又は使用基準改正の要請書に添付すべき資料 |
| 資料の種類 |
指定を要請
する場合 |
使用基準改正
を要請する場合 |
1.資料概要
2.起源又は発見の経緯及び外国における使用状況に関する資料
(1)起源又は発見の経緯
(2)外国における使用状況
3.物理化学的性質及び成分規格に関する資料
(1)名称
(2)構造式又は示性式
(3)分子式及び分子量
(4)含量規格
(5)製造方法
(6)性状
(7)確認試験
(8)示性値
(9)純度試験
(10)乾燥減量、強熱減量又は水分
(11)強熱残留物(強熱残分)
(12)定量法
(13)食品添加物の安定性
(14)食品中の食品添加物の分析法
(15)成分規格案の設定根拠
4.有効性に関する資料
(1)食品添加物としての有効性及び他の同種の添加物との効果の比較
(2)食品中での安定性
(3)食品中の栄養成分に及ぼす影響
5.安全性に関する資料
(1)毒性に関する資料
(1)28日反復投与毒性試験
(2)90日反復投与毒性試験
(3)1年間反復投与毒性試験
(4)繁殖試験
(5)催奇形性試験
(6)発がん性試験
(7)1年間反復投与毒性/発がん性併合試験
(8)抗原性試験
(9)変異原性試験
(10)一般薬理試験
(2)体内動態に関する資料
(3)食品添加物の1日摂取量に関する資料
6.使用基準案に関する資料 |
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○
○ |
○
△
○
△
△
△
△
△
△
△
△
△
△
△
△
△
△
△
○
△
△
△
△
△
△
△
△
△
△
△
△
△
○
○ |
| (注) | ○印は添付すべき資料。△印は新たな知見がある場合等必要な場合に添付すべき資料を示す。 |
| 表2 | 食品添加物が食品内又は消化管内で分解して食品常在成分となることを確認する場合の検討事項 |
| 1. | 食品添加物の通常の使用条件下で、当該物質が容易に食品内又は消化管内で分解して食品常在成分と同一物質になること。
|
| 2. | 食品内又は消化管内での分解に関わる主要な因子(pH、酵素等)が明らかであること。
|
| 3. | 食品添加物の通常の使用条件下で適正な量を使用した場合、当該食品添加物の体内への吸収が食品成分と同程度であり、他の栄養成分の吸収を阻害しないこと。
|
| 4. | 摂取された食品添加物の未加水分解物又は部分加水分解物が大量に糞便中に排泄されないこと。更に、未加水分解物又は部分加水分解物が生体組織中に蓄積しないこと。
|
| 5. | 食品添加物を使用した食品を摂取したとき、当該食品の主成分の過剰摂取の問題が起きないこと。 |
様式1
年 月 日
厚生大臣殿
住所(法人にあっては、主たる事務所の所在地)
氏名(法人にあっては、名称及び代表者の氏名) |
印 |
食品衛生法第6条の規定により人の健康を損なうおそれがないものとして下記品目を指定されるよう要請します。
記
( 品名 )
(注意)
| 1. | 用紙の大きさは、日本工業規格A4とすること。 |
| 2. | 字は、墨、インク等を用い、邦文にあっては楷書ではっきり書くこと。 |
| 3. | 要請者が外国に在住する場合には、国内連絡先を記載すること。なお、印を署名に代えることができる。 |
様式2
年 月 日
厚生大臣殿
住所(法人にあっては、主たる事務所の所在地)
氏名(法人にあっては、名称及び代表者の氏名) |
印 |
食品衛生法第7条第1項の規定による食品添加物使用基準の一部を下記のとおり改正されるよう要請します。
記
(品名及び使用基準改正案)
(注意)
| 1. | 用紙の大きさは、日本工業規格A4とすること。 |
| 2. | 字は、墨、インク等を用い、邦文にあっては楷書ではっきり書くこと。 |
| 3. | 要請者が外国に在住する場合には、国内連絡先を記載すること。なお、印を署名に代えることができる。 |
V 安全性に関する試験の標準的実施方法
| [1] | 28日間反復投与毒性試験
本試験は、げっ歯類及び非げっ歯類を用いて被験物質を28日間繰り返し投与したときに生じる毒性影響についての情報を提供し、また、あわせて1年間反復投与毒性試験等の用量設定の為の情報を提供することを目的とする。 |
| 1. | 動物種及び性
げっ歯類1種(通常、ラットが用いられる。)及び非げっ歯類1種(通常、イヌが用いられる。)について実施する。雌雄の動物を原則として同数用いる。
げっ歯類(ラット、マウス)については、離乳後、馴化期間を経てできるだけ早い時期の同一週齢の動物、通常5〜6週齢の動物を用い、非げっ歯類(イヌ)については、4〜6カ月齢の動物を用いる。
|
| 2. | 動物数
試験に用いる動物数は、げっ歯類では1群当たり雌雄各5匹以上、非げっ歯類では1群当たり雌雄各4匹以上とする。各群への動物の割り付けには、体重層別等による適切な無作為抽出法を用いる。
なお、最終的に試験結果の評価を行うのに十分な動物数が確保できていなければならない。
|
| 3. | 投与経路
被験物質の投与経路は、経口投与とし、通常、混餌投与又は飲水投与により行う。ただし、被験物質が飼料(飲水)中で不安定である場合、飼料(飲水)中の被験物質の分析が困難である場合、又は飼料(飲水)が忌避される場合等混餌(飲水)投与が困難な場合には強制投与を行うことも差し支えない。また、一定の条件下において、被験物質をマイクロカプセルに封入して投与しても差し支えない。
|
| 4. | 投与期間
被験物質の投与は、原則として週7日、28日間とする。
|
| 5. | 用量段階及び対照群
| (1) | 用量段階
対照群の他に少なくとも3段階の投与群を設ける。
用量段階は被験物質の毒性の全容を明らかにし、無毒性量(NOAEL)を推定することができるように設定する。したがって、最高用量は毒性影響が認められる用量、最低用量は何ら毒性影響が認められない用量とし、かつ用量反応関係がみられるように各用量段階を設定する。また、用量設定の根拠を示すこと。
なお、混餌投与の場合は、栄養障害が起こらないよう十分配慮し、通常、飼料添加濃度5%(W/W)を超える投与量で実施する必要はない。また、強制投与の場合には、通常、技術的に投与できる最大量又は1000mg/kgで何ら毒性影響が認められない場合は、それ以上の投与量で実施する必要はない。
|
| (2) | 対照群
対照群としては、被験物質の投与を行わないこと以外、すべての点で被験物質投与群と同一条件の群を設ける。
被験物質の投与に媒体等を使用する場合には、投与媒体量の最も多い用量 群と同量の投与を行うこと。毒性に関する情報が十分に得られていない媒体 等を使用する場合には、さらに無処置対照群を加える。 |
|
| 6. | 観察及び検査
次の(1)〜(6)の項目について実施する。
| (1) | 一般状態、体重、摂餌(摂水)量
全試験動物について、一般状態を毎日観察し、定期的に体重及び摂餌量(飲水投与の場合は摂水量)を測定する。一般状態の観察においては、次の項目についての観察及び検査を行う。
外観(被毛、皮膚、眼・眼球、耳、鼻、口、肛門周囲及び粘膜等の変化、並びに衰弱、肥満・るいそう、腹部膨満等)、体位・姿勢(腹臥位、はいずり姿勢、円背位等)、意識・態度(興奮、攻撃性、不活発、鎮静、嗜眠等)、行動(探索行動の変化、身づくろいの変化、自発行動の変化、歩行の異常等)、神経系(振戦、けい攣、筋収縮性、各種の反射機能等)、体温、呼吸状態、排泄状態
なお、体重及び摂餌(摂水)量の測定の頻度は、通常、次のとおりである。
|
| (2) | 血液検査
げっ歯類では剖検前に少なくとも1回、非げっ歯類では投与開始前、剖検前に採血して検査する。
検査は、通常、次の項目について検査されるが、そのほか試験ごとに適切な項目を追加選定して行うことが望ましい。なお、検査項目及び検査方法は、国際的に広く採用されているものを考慮に入れて選定するべきである。
血液学的検査:赤血球数、白血球数、血液像(白血球型別百分率)、血小板数、血色素、ヘマトクリット、その他必要に応じて網状赤血球、凝固能(プロトロンビン時間、活性化部分トロンボプラスチン時間)等血液生化学的検査:血清(血漿)総蛋白、アルブミン、A/G比、蛋白分画*、ブドウ糖、コレステロール、トリグリセリド、ビリルビン、尿素窒素、クレアチニン、トランスアミナーゼ[AST(GOT),ALT(GPT)]、γ−GTP、アルカリホスファターゼ、電解質(ナトリウム、カリウム、塩素、カルシウム、無機リン等)等
| *: | 血清総蛋白に異常が認められた場合は免疫グロブリン分画(IgG、IgM、IgE、IgA)についても検査する。 |
|
| (3) | 尿検査
全試験動物について、投与開始前及び剖検前に尿検査を行う。
検査は、通常、次の項目について実施される。
尿量、pH、蛋白、糖、ケトン体、ビリルビン、ウロビリノーゲン、潜血、沈渣、比重又は浸透圧、電解質(ナトリウム、カリウム等)
|
| (4) | 眼科学的検査
げっ歯類については、最高用量群と対照群について剖検前に、非げっ歯類については、全試験動物について投与開始前及び剖検前に眼科学的検査を行う。げっ歯類の最高用量群に変化が認められた場合は、その他の用量群についても検査する。
検査は、通常、肉眼的及び検眼鏡的に行い、前眼房・中間透光体・眼底のそれぞれについて実施される。
|
| (5) | その他の機能検査
必要に応じて、心電図、腎機能、感覚機能検査や自発運動量測定、更により高次の行動変化等の検査を実施する。
|
| (6) | 剖検及び病理組織学的検査
| (1) | 投与期間中の死亡例は速やかに剖検し、器官・組織の肉眼的観察を行うほか、必要に応じ、器官の重量測定、病理組織学的検査を行い、死因とその時点での毒性変化の程度とを明らかにするよう努める。 |
| (2) | 投与期間中に死に瀕した例は、死を待つより殺処分を行う方が多くの知見が得られるので、速やかに屠殺、剖検し、(1)と同様の観察、検査を行い、瀕死状態となった理由とその時点での毒性変化の程度とを明らかにするよう努める。 |
| (3) | 投与期間終了時のすべての生存例は、諸検査等のための採血、採尿を行った後、屠殺、剖検し、器官・組織の肉眼的観察を行い、器官について重量測定を行う。重量測定は、通常、以下の器官について行われる。
心臓、肝臓、脾臓、腎臓、副腎、前立腺、精巣、精嚢、卵巣、子宮、脳、下垂体、唾液腺、胸腺、肺、甲状腺・上皮小体
病理組織学的検査は、非げっ歯類では全試験動物の、げっ歯類では少なくとも最高用量群と対照群の動物の組織について行う。通常、以下の器官の組織について行われるが、肉眼的観察所見等からの判断によって適宜追加する。
皮膚、乳腺、リンパ節(頚部リンパ節、腸間膜リンパ節等)、大動脈、唾液腺、骨及び骨髄(胸骨、大腿骨)、胸腺、気管、肺及び気管支、心臓、甲状腺及び上皮小体、舌、食道、胃、十二指腸、小腸(空腸、回腸)、大腸(盲腸、結腸、直腸)、肝臓及び胆嚢、膵臓、脾臓、腎臓、副腎、膀胱、精嚢、前立腺、精巣・精巣上体、卵巣及び卵管、子宮、膣、脳、下垂体、坐骨神経、骨格筋、脊髄、鼻腔(鼻甲介)、眼球及びその附属器、ジンバル腺、そのほか肉眼的に変化が認められた器官・組織
なお、これらの組織のうち、免疫系、神経系及び性腺に対する影響を見るためには、特に次の点を考慮する。
免疫系:胸腺、脾臓とリンパ節/リンパ様組織については必要により免疫組織化学検査を行い、免疫毒性が疑われた時は、更に、骨髄構成細胞比、脾リンパ球組成、NK細胞活性についても検査することが望ましい。
神経系:被験物質の投与によると考えられる神経症状の発現がみられた場合、一部の動物に対して潅流固定法により固定を行い、前脳、大脳中央部、中脳、小脳、橋、延髄、脊髄(頚、胸、腰部)、背根神経節、背根、腹根、近位坐骨神経、腓腹神経、腓腹筋について神経病理学的検査を行う。
性腺:精巣は、通常、ブアン固定を用いて検索する。
また、げっ歯類の他の用量群についても、肉眼的に変化の認められた器官のある場合、あるいは高用量群での所見から考えて、必要と考えられる場合には、当該器官・組織についてその用量群の全試験動物について病理組織学的検査を行う。
なお、上述の場合のほかにあっても、げっ歯類において、全試験動物について病理組織学的検査を行うことは、評価の助けになる。 |
|
|
| [2] | 90日間反復投与毒性試験
本試験は、げっ歯類及び非げっ歯類を用いて被験物質を90日間以上繰り返し投与したときに生じる毒性変化についての情報を与える。また、発がん性及び1年間反復投与毒性/発がん性併合試験等の用量設定のための情報を提供することを目的とする。 |
| 1. | 動物種及び性
げっ歯類1種(通常、ラットが用いられる。)及び非げっ歯類1種(通常、イヌが用いられる。)について実施する。雌雄の動物を原則として同数用いる。
げっ歯類(ラット、マウス)については、離乳後、馴化期間を経てできるだけ早い時期の同一週齢の動物、通常5〜6週齢の動物を用い、非げっ歯類(イヌ)については、4〜6カ月齢の動物を用いる。
|
| 2. | 動物数
試験に用いる動物数は、げっ歯類では1群当たり雌雄各10匹以上、非げっ歯類では1群当たり雌雄各4匹以上とする。各群への動物の割り付けには、体重層別等による適切な無作為抽出法を用いる。なお、中間屠殺を予定する場合は、そのために必要な数を追加すること。
なお、最終的に試験結果の評価を行うのに十分な動物数が確保できていなければならない。
|
| 3. | 投与経路
被験物質の投与経路は、経口投与とし、通常、混餌投与又は飲水投与により行う。ただし、被験物質が飼料(飲水)中で不安定である場合、飼料(飲水)中の被験物質の分析が困難である場合、又は飼料(飲水)が忌避される場合等混餌(飲水)投与が困難な場合には強制投与を行うことも差し支えない。また、一定の条件下において、被験物質をマイクロカプセルに封入して投与しても差し支えない
|
| 4. | 投与期間
被験物質の投与は、原則として週7日、90日以上とする。
|
| 5. | 用量段階及び対照群
| (1) | 用量段階
対照群の他に少なくとも3段階の投与群を設ける。
用量段階は被験物質の毒性の全容を明らかにし、無毒性量(NOAEL)を推定することができるように設定する。したがって、最高用量は多数例の死亡を起こすことなく毒性影響が認められる用量、最低用量は何ら毒性影響が認められない用量とし、かつ用量反応関係がみられるように各用量段階を設定する。また、用量設定の根拠を示すこと。
なお、混餌投与の場合は、栄養障害が起こらないよう十分配慮し、通常、飼料添加濃度5%(W/W)を超える投与量で実施する必要はない。また、強制投与の場合には、通常、技術的に投与できる最大量又は1000mg/kgで何ら毒性影響が認められない場合は、それ以上の投与量で実施する必要はない。
|
| (2) | 対照群
対照群としては、被験物質の投与を行わないこと以外、すべての点で被験物質投与群と同一条件の群を設ける。
被験物質の投与に媒体等を使用する場合には、投与媒体量の最も多い用量群と同量の投与を行うこと。毒性に関する情報が十分に得られていない媒体等を使用する場合には、さらに無処置対照群を加える。 |
|
| 6. | 観察及び検査
次の(1)〜(6)の項目について実施する。
| (1) | 一般状態、体重、摂餌(摂水)量
全試験動物について、一般状態を毎日観察し、定期的に体重及び摂餌量(飲水投与の場合は摂水量)を測定する。一般状態の観察においては、次の項目についての観察及び検査を行う。
外観(被毛、皮膚、眼・眼球、耳、鼻、口、肛門周囲及び粘膜等の変化、並びに衰弱、肥満・るいそう、腹部膨満等)、体位・姿勢(腹臥位、はいずり姿勢、円背位等)、意識・態度(興奮、攻撃性、不活発、鎮静、嗜眠等)、行動(探索行動の変化、身づくろいの変化、自発行動の変化、歩行の異常等)、神経系(振戦、けい攣、筋収縮性、各種の反射機能等)、体温、呼吸状態、排泄状態
なお、体重及び摂餌(摂水)量の測定の頻度は、通常、次のとおりである。
| 摂餌(摂水)量: | 投与開始前及び投与開始後少なくとも週1回。 |
|
| (2) | 血液検査
げっ歯類では剖検前に少なくとも1回、非げっ歯類では投与開始前、剖検前に採血して検査する。検査は原則として全試験動物について行うべきであるが、げっ歯類については、実施上の理由から、各群の一部の動物に限っても差し支えない。
検査は、通常、次の項目について検査されるが、そのほか試験ごとに適切な項目を追加選定して行うことが望ましい。なお、検査項目及び検査方法は、国際的に広く採用されているものを考慮に入れて選定するべきである。
血液学的検査:赤血球数、白血球数、血液像(白血球型別百分率)、血小板数、血色素、ヘマトクリット、その他必要に応じて網状赤血球、凝固能(プロトロンビン時間、活性化部分トロンボプラスチン時間)等
血液生化学的検査:血清(血漿)総蛋白、アルブミン、A/G比、蛋白分画*、ブドウ糖、コレステロール、トリグリセリド、ビリルビン、尿素窒素、クレアチニン、トランスアミナーゼ[AST(GOT),ALT(GPT)]、γ−GTP、アルカリホスファターゼ、電解質(ナトリウム、カリウム、塩素、カルシウム、無機リン等)等
|
| (3) | 尿検査
げっ歯類では雌雄各群ごとに一定数の動物(少くとも5匹)を選び、非げっ歯類では各群の全試験動物について、血液検査と同時期に尿検査を行う。
検査は、通常、次の項目について実施される。
尿量、pH、蛋白、糖、ケトン体、ビリルビン、ウロビリノーゲン、潜血、沈渣、比重又は浸透圧、電解質(ナトリウム、カリウム等)
|
| (4) | 眼科学的検査
げっ歯類では必要に応じて、投与期間中少なくとも1回(終了時)、雌雄各群ごとに一定数の動物(少くとも5匹)を選び、眼科学的検査を行う。また、非げっ歯類では投与開始前、投与期間中少なくとも1回及び剖検前に、各群の全試験動物について眼科学的検査を行う。
検査は、通常、肉眼的及び検眼鏡的に行い、前眼房・中間透光体・眼底のそれぞれについて実施される。
|
| (5) | その他の機能検査
必要に応じて、心電図、腎機能、感覚機能検査や自発運動量測定、更により高次の行動変化等の検査を行う。
|
| (6) | 剖検及び病理組織学的検査
| (1) | 投与期間中の死亡例は速やかに剖検し、器官・組織の肉眼的観察を行うほか、必要に応じ、器官の重量測定、病理組織学的検査を行い、死因とその時点での毒性変化の程度とを明らかにするよう努める。 |
| (2) | 投与期間中に死に瀕した例は、死を待つより殺処分を行う方が多くの知見が得られるので、速やかに屠殺、剖検し、(1)と同様の観察、検査を行い、瀕死状態となった理由とその時点での毒性変化の程度とを明らかにするよう努める。 |
| (3) | 投与期間終了時のすべての生存例は、諸検査等のための採血、採尿を行った後、屠殺、剖検し、器官・組織の肉眼的観察を行い、器官について重量測定を行う。重量測定は、通常、以下の器官について行われる。
心臓、肝臓、脾臓、腎臓、副腎、前立腺、精巣、精嚢、卵巣、子宮、脳、下垂体、唾液腺、胸腺、肺、甲状腺・上皮小体
病理組織学的検査は、非げっ歯類では全試験動物の、げっ歯類では少なくとも最高用量群と対照群の動物の組織について行う。通常、以下の器官の組織について行われるが、肉眼的観察所見等からの判断によって適宜追加する。
皮膚、乳腺、リンパ節(頚部リンパ節、腸間膜リンパ節等)、大動脈、唾液腺、骨及び骨髄(胸骨、大腿骨)、胸腺、気管、肺及び気管支、心臓、甲状腺及び上皮小体、舌、食道、胃、十二指腸、小腸(空腸、回腸)、大腸(盲腸、結腸、直腸)、肝臓及び胆嚢、膵臓、脾臓、腎臓、副腎、膀胱、精嚢、前立腺、精巣・精巣上体、卵巣及び卵管、子宮、膣、脳、下垂体、坐骨神経、骨格筋、脊髄、鼻腔(鼻甲介)、眼球及びその附属器、ジンバル腺、そのほか肉眼的に変化が認められた器官・組織
なお、これらの組織のうち、免疫系、神経系及び性腺に対する影響を見るためには、特に次の点を考慮する。
免疫系:胸腺、脾臓とリンパ節/リンパ様組織については必要により免疫組織化学検査を行い、免疫毒性が疑われた時は、更に、骨髄構成細胞比、脾リンパ球組成、NK細胞活性についても検査することが望ましい。
神経系:被験物質の投与によると考えられる神経症状の発現が見られた場合、一部の動物に対して潅流固定法により固定を行い、前脳、大脳中央部、中脳、小脳、橋、延髄、脊髄(頚、胸、腰部)、背根神経節、背根、腹根、近位坐骨神経、腓腹神経、腓腹筋について神経病理学的検査を行う。
性腺:精巣は、通常、ブアン固定を用いて検索する。
また、げっ歯類の他の用量群についても、肉眼的に変化の認められた器官のある場合、あるいは高用量群での所見から考えて、必要と考えられる場合には、当該器官・組織についてその用量群の全試験動物について病理組織学的検査を行う。
なお、上述の場合のほかにあっても、げっ歯類において、全試験動物について病理組織学的検査を行うことは、評価の助けになる。 |
|
|
| [3] | 1年間反復投与毒性試験
本試験は、げっ歯類及び非げっ歯類を用いて被験物質を長期間にわたって繰り返し投与したとき、明らかな毒性変化を惹起する用量とその変化の内容及び毒性変化の認められない用量を求めることを目的とする。 |
| 1. | 動物種及び性
げっ歯類1種(通常、ラットが用いられる。)及び非げっ歯類1種(通常、イヌが用いられる。)について実施する。雌雄の動物を原則として同数用いる。げっ歯類(ラット、マウス)については、離乳後、馴化期間を経てできるだけ早い時期の同一週齢の動物、通常5〜6週齢の動物を用い、非げっ歯類(イヌ)については4〜6カ月齢の動物を用いる。
|
| 2. | 動物数
試験に用いる動物数は、げっ歯類では1群当たり雌雄各20匹以上、非げっ歯類では1群当たり雌雄各4匹以上とする。各群への動物の割り付けには、体重層別等による適切な無作為抽出法を用いる。なお、中間屠殺を予定する場合は、そのために必要な数を追加すること。
なお、最終的には、試験結果の評価を行うのに十分な動物数が確保できていなければならない。
|
| 3. | 投与経路
被験物質の投与経路は、経口投与とし、通常、混餌投与又は飲水投与により行う。ただし、被験物質が飼料(飲水)中で不安定である場合、飼料(飲水)中の被験物質の分析が困難である場合、又は飼料(飲水)が忌避される場合等混餌(飲水)投与が困難な場合には強制投与を行うことも差し支えない。また、一定の条件下において、被験物質をマイクロカプセルに封入して投与しても差し支えない。
|
| 4. | 投与期間
被験物質の投与は、原則として週7日、1年以上とする。
|
| 5. | 用量段階及び対照群
| (1) | 用量段階
対照群の他に少なくとも3段階の投与群を設ける。
用量段階は被験物質の毒性の全容を明らかにし、無毒性量(NOAEL)を推定することができるように設定する。したがって、最高用量は多数例の死亡を起こすことなく毒性影響が認められる用量、最低用量は何ら毒性影響が認められない用量とし、かつ用量反応関係がみられるように各用量段階を 設定する。用量設定にあたっては、28日又は90日間反復投与毒性試験の結果等を参考とすること。また、用量設定の根拠を示すこと。
なお、混餌投与の場合は、栄養障害が起こらないよう十分配慮し、通常、飼料添加濃度5%(W/W)を超える投与量で実施する必要はない。また、強制投与の場合には、通常、技術的に投与できる最大量又は1000mg/kgで何ら毒性影響が認められない場合は、それ以上の投与量で実施する必要はない。
|
| (2) | 対照群
対照群としては、被験物質の投与を行わないこと以外、すべての点で被験物質投与群と同一条件の群を設ける。
被験物質の投与に媒体等を使用する場合には、投与媒体量の最も多い用量群と同量の投与を行うこと。毒性に関する情報が十分に得られていない媒体等を使用する場合には、さらに無処置対照群を加える。 |
|
| 6. | 観察及び検査
次の(1)〜(6)の項目について実施する。
| (1) | 一般状態、体重、摂餌(摂水)量
全試験動物について、一般状態を毎日観察し、定期的に体重及び摂餌量(飲水投与の場合は摂水量)を測定する。一般状態の観察においては、次の項目についての観察及び検査を行う。
外観(被毛、皮膚、眼・眼球、耳、鼻、口、肛門周囲及び粘膜等の変化、並びに衰弱、肥満・るいそう、腹部膨満等)、体位・姿勢(腹臥位、はいずり姿勢、円背位等)、意識・態度(興奮、攻撃性、不活発、鎮静、嗜眠等)、行動(探索行動の変化、身づくろいの変化、自発行動の変化、歩行の異常等)、 神経系(振戦、けい攣、筋収縮性、各種の反射機能等)、体温、呼吸状態、排泄状態
なお、体重及び摂餌(摂水)量の測定の頻度は、通常、次のとおりである。
| 体重: | 投与開始前及び投与開始後3カ月までは少なくとも週1回、その後は少なくとも4週に1回。 |
| 摂餌(摂水)量: | 投与開始前及び投与開始後3カ月までは少なくとも週1回、その後は少なくとも4週に1回。 |
なお、この測定はケージごとに行ってもよい。
|
| (2) | 血液検査
げっ歯類では剖検前に、非げっ歯類では投与開始前、投与開始後3カ月ごと及び試験終了時に採血して検査する。検査は原則として全試験動物について行うべきであるが、げっ歯類については、実施上の理由から、各群の一部の動物に限っても差し支えない。
検査は、通常、次の項目について検査されるが、そのほか試験ごとに適切な項目を追加選定して行うことが望ましい。なお、検査項目及び検査方法は、国際的に広く採用されているものを考慮に入れて選定するべきである。
血液学的検査:赤血球数、白血球数、血液像(白血球型別百分率)、血小板数、血色素、ヘマトクリット、その他必要に応じて網状赤血球、凝固能(プロトロンビン時間、活性化部分トロンボプラスチン時間)等
血液生化学的検査:血清(血漿)総蛋白、アルブミン、A/G比、蛋白分画、ブドウ糖、コレステロール、トリグリセリド、ビリルビン、尿素窒素、クレアチニン、トランスアミナーゼ[AST(GOT),ALT(GPT)]、γ−GTP、アルカリホスファターゼ、電解質(ナトリウム、カリウム、塩素、カルシウム、無機リン等)等
|
| (3) | 尿検査
げっ歯類では各群ごとに一定数の動物(少くとも10匹)を選び、非げっ歯類では各群の全試験動物について、血液検査と同時期に尿検査を行う。
検査は、通常、次の項目について実施される。
尿量、pH、蛋白、糖、ケトン体、ビリルビン、ウロビリノーゲン、潜血、沈渣、比重又は浸透圧、電解質(ナトリウム、カリウム等)
|
| (4) | 眼科学的検査
げっ歯類では必要に応じて、投与期間中少なくとも1回(終了時)、雌雄各群ごとに一定数の動物(少くとも10匹)を選び、眼科学的検査を行う。また、非げっ歯類では投与開始前及び投与期間中少なくとも1回及び剖検前に、各群の全試験動物について眼科学的検査を行う。
検査は、通常、肉眼的及び検眼鏡的に行い、前眼房・中間透光体・眼底のそれぞれについて実施される。
|
| (5) | その他の機能検査
必要に応じて、心電図、視覚、聴覚、腎機能等の検査を行う。
|
| (6) | 剖検及び病理組織学的検査
| (1) | 投与期間中の死亡例は速やかに剖検し、器官・組織の肉眼的観察を行うほか、必要に応じ、器官の重量測定、病理組織学的検査を行い、死因とその時点での毒性変化の程度とを明らかにするよう努める。 |
| (2) | 投与期間中に死に瀕した例は、死を待つより殺処分を行う方が多くの知見が得られるので、速やかに屠殺、剖検し、(1)と同様の観察、検査を行い、瀕死状態となった理由とその時点での毒性変化の程度とを明らかにするよう努める。 |
| (3) | 投与期間終了時のすべての生存例は、諸検査等のための採血、採尿を行った後、屠殺、剖検し、器官・組織の肉眼的観察を行い、器官について重量測定を行う。重量測定は、通常、以下の器官について行われる。
心臓、肝臓、脾臓、腎臓、副腎、前立腺、精巣、精嚢、卵巣、子宮、脳、下垂体、唾液腺、胸腺、肺、甲状腺・上皮小体
病理組織学的検査は、非げっ歯類では全例の、げっ歯類では少なくとも最高用量群と対照群の組織について行う。通例、以下の器官の組織について行われるが、肉眼的所見等からの判断によって適宜追加する。
皮膚、乳腺、リンパ節(頚部リンパ節、腸間膜リンパ節等)、大動脈、唾液腺、骨及び骨髄(胸骨、大腿骨)、胸腺、気管、肺及び気管支、心臓、甲状腺及び上皮小体、舌、食道、胃、十二指腸、小腸(空腸、回腸)、大腸(盲腸、結腸、直腸)、肝臓及び胆嚢、膵臓、脾臓、腎臓、副腎、膀胱、精嚢、前立腺、精巣・精巣上体、卵巣及び卵管、子宮、膣、脳、下垂体、坐骨神経、骨格筋、脊髄、鼻腔(鼻甲介)、眼球及びその附属器、ジンバル腺、そのほか肉眼的に変化が認められた器官・組織
また、げっ歯類の他の用量群についても、肉眼的に変化の認められた器官のある場合、あるいは高用量群での所見から考えて、必要と考えられる場合には、当該器官・組織についてその用量群の全試験動物について病理組織学的検査を行う。
なお、上述の場合のほかにあっても、げっ歯類において、全試験動物について病理組織学的検査を行うことは、評価の助けになる。 |
|
|
| [4] | 繁殖試験
本試験は、被験物質を二世代(第一世代(F0)及び第二世代(F1))にわたって投与し、発情、交尾、受胎、分娩、哺育等の生殖機能、離乳及び出産後の新生児の生育に及ぼす影響に関する情報を得ることを目的とする。また、本試験から、胎児の死亡及び奇形発生に関する予備的な情報が得られ、関連する試験を実施するにあたっての参考に資することができる。 |
| 1. | 動物種
げっ歯類1種以上(通常、ラットが用いられる。)について実施する。雌雄の動物を原則として同数用いる。なお、使用する雌は未経産のものでなければならない。
種及び系統の選択に当たっては、産児数の少ない系統は避け、一般毒性試験あるいは繁殖試験に繁用されているものを選ぶ。
ラットについては、1週間以上の馴化期間を経た動物で雄は通常5〜7週齢動物を、雌は通常8〜10週齢の動物を用いる。
|
| 2. | 動物数
試験に用いる動物数は、ラットでは交配時点に1群あたり20組以上が得ら れるだけの数の雌雄とする。各群への動物の割り付けには、体重層別等による 適切な無作為抽出法を用いる。
ラット以外の動物を用いる場合には、評価に耐える知見が得られると期待さ れる動物数とする。
|
| 3. | 投与経路
被験物質の投与経路は、経口投与とし、通常、混餌投与又は飲水投与により行う。ただし、被験物質が飼料(飲水)中で不安定である場合、飼料(飲水)中からの被験物質の分析が困難である場合又は飼料(飲水)が忌避される場合等混餌(飲水)投与が困難な場合には強制投与を行うことも差し支えない。
この場合、各動物に対する投与量は各週の個体体重に基づいて計算する。妊娠期の雌動物については、妊娠0日及び妊娠6日の体重に基づいて投与量を決定してもよい。
|
| 4. | 投与期間
被験物質の投与期間は、次のとおりである。
| (1) | 第一世代(F0)の雌動物
8〜10週齢から投与を開始し、2週間以上投与した後、交配する。交配期間、妊娠期間及び哺育期間を通じ、出生児(F1)が離乳するまで連日投与する。
|
| (2) | 第一世代(F0)の雄動物
5〜7週齢から投与を開始し、8週間以上投与した後、交配する。交配期間中も連日投与する。
|
| (3) | 第二世代(F1)
離乳時から投与を開始し、雌動物については次の出生児(F2)が離乳するまで、雄動物については交配が終了するまで投与する。 |
|
| 5. | 用量段階と対照群
| (1) | 用量段階
対照群の他に少なくとも3段階の投与群を設ける。
用量段階は被験物質の毒性の全容を明らかにし、無毒性量(NOAEL)を推定することができるように設定する。したがって、最高用量は母動物に体重増加抑制等何らかの毒性影響が認められるが死には至らしめない用量、最低用量は親動物及び子動物のいずれにも何ら毒性影響が認められない用量とし、かつ用量反応関係がみられるように各用量段階を設定する。また、用量設定の根拠を示すこと。
なお、混餌投与の場合は、栄養障害が起こらないよう十分配慮し、通常、飼料添加濃度5%(W/W)を超える投与量で実施する必要はない。また、強制投与の場合には、通常、1000mg/kg又は技術的に投与できる最大量で何ら毒性影響が認められない場合は、それ以上の投与量で実施する必要はない。
|
| (2) | 対照群
対照群としては、被験物質の投与を行わない以外すべての点で被験物質投与群と同一条件の群を設ける。
被験物質の投与に媒体等を使用する場合には、投与媒体量の最も多い用量群と同量の投与を行うこと。毒性に関する情報が十分に得られていない媒体等を使用する場合には、さらに無処置対照群を加える。 |
|
| 6. | 交配並びに第二世代(F1)の選抜
| (1) | 第一世代(F0)
同じ用量群の雌雄を1対1で同居させ交尾が確認されるまで交配させる。同居期間は3週間を限度とする。
雌動物については、毎朝膣垢内の精子又は膣栓の検査を行い、交尾の有無を確認し、精子又は膣栓が認められた日を妊娠0日とする。
|
| (2) | 第二世代(F1)
| (1) | 同腹児数の調整
生後4日に、各同腹児が雄雌各4匹になるように、余分な新生児を無作為に取り除く。なお、1腹当たり雌雄各4匹に調整することができない場合には、総数8匹(例えば雄5匹と雌3匹)に調整すればよい。また、同腹児の匹数が8匹以下の場合には調整を行わない。 |
| (2) | 交配用(F1)の選抜
F1の離乳時に、各群ともできるだけ多くの母動物から、雌雄各20〜25匹(通常は各母動物から雌雄各1〜2匹)のF1を交配用として選抜する。 |
| (3) | 交配
雌雄とも10〜13週齢で同腹児の交配を避けて第一世代と同様に行う。 |
|
|
| 7. | 観察及び検査
次の(1)〜(5)の項目について実施する。
必要に応じて親動物、子動物の免疫系及び神経系に対する影響について検査を行う。
| (1) | 一般状態
全試験動物(F0,F1,F2)について、一般状態及び妊娠・分娩状態を毎日観察する。
F0及び離乳後のF1動物の一般状態としては、生死、外観のほか、興奮、けいれん、鎮静、歩行異常等を観察する。妊娠・分娩状態としては、流産・早産、分娩遅延等を観察する。
新生児については、発達の身体的指標(耳介の展開、切歯の萌芽、体毛の出現、開眼、生殖器の発育等)及び発達の機能的指標(正向反射、運動性、聴覚性驚愕反応等)等を観察又は検査する。
|
| (2) | 体重、摂餌(摂水)量
F0動物及び交配用F1動物について、定期的に体重、摂餌量及び摂水量を測定する。体重及び摂餌(摂水)量の測定の頻度は、通常、次のとおりである。
| 体重: | 投与開始前及び投与開始後は少なくとも週1回。 |
| 摂餌(摂水)量: | 投与開始前及び投与開始後は少なくとも週1回。 |
|
| (3) | 妊娠、出産について
交尾動物数、妊娠動物数(及び妊娠させ得た雄数)、出産母体数及び離乳児数をもとにして、以下のパラメータを算出する。
交尾率=(交尾動物数/交配に用いた動物数)×100
妊娠率=(妊娠動物数/交尾した雌動物数)×100
出産率=(生児出産雌数/妊娠雌数)×100
離乳時の生存率=(離乳時の生存児数/生後4日に調整した児数)×100
交尾が成立しなかった雌雄動物については、他の雄又は雌動物との再交配、発情周期及び精子形成の検査及び生殖器の組織学的検査等を行う。また、妊娠が成立しなかった場合の精子形成検査は、屠殺時における精子の数、運動性及び形態等に関して実施すること。
|
| (4) | 新生児
母動物ごとに、出産後できる限り早い時期に産児数、死児数、生存児数を数え外表異常の有無及び性別を調べる。
また、死児及び生後4日に屠殺したF1については、剖検を行う。生存児については、出産直後又は出産後の早い時期及び生後4日、7日(任意)、14日、21日にそれぞれ匹数を調べ、体重を測定する。
|
| (5) | 剖検
| (1) | F0動物及び交配に用いたF1動物は、それぞれの出生児の離乳後速やかに、交配用に選抜されなかったF1動物及びF2動物は離乳後速やかに屠殺、剖検し、特に生殖器系の器官に注意を払いながら、肉眼的観察を行う。試験期間中の死亡例又は試験期間中に死に瀕した例は速やかに屠殺、剖検し、同様の観察を行う。 |
| (2) | 被験物質投与の影響と考えられる肉眼的異常が認められた器官及び組織については、病理組織学的検査を行う。また、反復投与毒性試験の結果を参考にする。 |
|
|
| 8. | 結果の解析
| (1) | 得られた成績を表又は図で示し、考察を加える。表示方法としては、全群の成績を概括する総括表を作成するほか、群ごとに個々の動物のデータを記した表も作成し、必要に応じて参照できるようにしておく。
|
| (2) | データの統計学的分析に際しては、離乳期までは各児を独立した標本として扱わず、1腹児を標本単位とすることが望ましい。
|
| (3) | 考察には、当該試験における親動物の生殖及び次世代の発生における無毒性量(NOAEL)についての見解を含めること。 |
|
| [5] | 催奇形性試験
本試験は、妊娠中の母動物が被験物質に暴露された場合の胎児の発生、発育に対する影響、特に催奇形性に関する情報を得ることを目的とする1)。 |
| 1. | 動物種
げっ歯類1種以上(通常、ラットが用いられる。)及び非げっ歯類(通常、ウサギが用いられる。)の合計2種以上について実施する。
|
| 2. | 動物数
一群あたりの雌雄動物数は、データの意味のある解釈が十分できる数とする 2)。
|
| 3. | 投与経路
被験物質の投与経路は、経口投与とし、強制投与する。1回で投与できない場合には、数回に分けて投与してもよいが、すべての投与を6時間以内に行うこと。各動物に対する投与量は、投与開始日の体重に基づいて計算する。血中濃度のデータや摂餌状況等より、一定の投与量が確保できることが確認できる場合には、飼料又は飲水に添加して投与を行っても差し支えない。
|
| 4. | 投与期間
被験物質の投与期間は、胎児の器官形性期を含む期間とし、連日投与を行う。
|
| 5. | 用量段階及び対照群
| (1) | 用量段階
対照群の他に少なくとも3段階の投与群を設ける。
用量段階は被験物質の毒性の全容を明らかにし、無毒性量(NOAEL)を推定することができるように設定する。したがって、最高用量は母動物に摂餌量の低下、体重増加の抑制等何らかの毒性影響が認められる量、最低用量は母動物、胎児のいずれにも何ら毒性影響が認められない用量とし、用量反応関係がみられるように各用量段階を設定する。また、用量設定の根拠を示すこと。
なお、混餌投与の場合には、栄養障害が起こらないよう十分配慮し、通常、飼料添加濃度5%(W/W)を超える投与量で実施する必要はない。また、強制投与の場合には、通常、1000mg/kg又は技術的に投与できる最大量で何ら毒性影響が認められない場合は、それ以上の投与量で実施する必要はない。
|
| (2) | 対照群
対照群としては、被験物質の投与を行わないこと以外、すべての点で被験物質投与群と同一条件の群を設ける。
被験物質の投与に溶媒等を使用する場合には、投与媒体量の最も多い用量群と同量の投与を行うこと。毒性に関する情報が十分に得られていない媒体等を使用する場合には、さらに無処置対照群を加える。 |
|
| 6. | 観察及び検査
次の(1)〜(2)の項目について実施する。
| (1) | 一般状態、体重、摂餌(摂水)量
母動物のすべてについて、一般状態及び妊娠状態を毎日観察し、体重、摂餌(摂水)量を定期的に測定する。
一般状態としては、生死、外観のほか、興奮、けいれん、鎮静、歩行異常等を観察する。妊娠状態としては、流産・早産等を観察する。
なお、体重及び摂餌(摂水)量の測定の頻度は、通常、次のとおりである。
| 体重: | 妊娠0日及び被験物質の投与開始後は、剖検まで毎日。 |
| 摂餌(摂水)量: | 少なくとも、投与開始前1回、投与期間中2回、投与終了後1回。 |
|
| (2) | 剖検
| (1) | 母動物
| ア) | 流産や早産の徴候を示した動物は速やかに屠殺し、子宮を摘出したのち、器官・組織の肉眼的観察を行うほか、必要に応じ、器官の重量測定、病理組織学的検査を行う。死亡動物及び死に瀕した動物についても、同様な検索を行う。摘出した子宮については、(2)の検査を行う。
|
| イ) | 出産予定日の前日にすべての動物を屠殺し、子宮を摘出したのち、すべての器官・組織の肉眼的観察を行うほか、必要に応じ、器官の重量の測定、病理組織学的検査を行う。摘出した子宮については、(2)の検査を行う。 |
|
| (2) | 胎児
| ア) | 母動物より摘出した子宮を切開し、胚死亡、胎児死亡及び生存胎児数を検査する。死亡胎児については、死亡時期を推定する根拠となる所見をできる限り記録する。ラット及びウサギでは黄体数も検査する。胎児の性別判定を行い、胎児体重を個別に測定、記録し、各群雌雄の平均胎児体重を算出する。 |
| イ) | 摘出したすべての胎児について外表異常の検査を行った後、ラットでは、同腹胎児の1/3〜1/2について骨格異常の検査を、残りについて内臓異常の検査を行い、ウサギでは、すべての胎児について内臓異常、骨格異常の検査を行う。 |
|
|
|
| 7. | 結果の解析
| (1) | 得られた成績を表又は図で示し、考察を加える。表示方法としては、全群の成績を概括する総括表を作成するほか、群ごとに個々の動物のデータを記した表も作成し、必要に応じて参照できるようにしておく。
|
| (2) | データの統計学的分析に際しては、1腹児を標本単位とすることが望ましい。
|
| (3) | 考察には、当該試験における親動物及び胎児に対する無毒性量(NOAEL)についての見解を含めること。 |
| 注1) | 本試験は、繁殖試験において交配に用いたF1をその出生児(F2a)の離乳後に屠殺せず、被験物質の投与を続けて、再度交配して得た胎児(F2b)を用いて催奇形性の観察を行うことができる。ただし、催奇形性試験を繁殖試験に併合して行なう場合は、次の2条件を満たした場合とする。
| (1) | 被験物の一般毒性や生体内動態等の知見からみて、催奇形性試験で最高投与量とすべき量による親動物と胎児への毒性影響が、繁殖試験の最高投与量による毒性影響に比べ、大きな差でないとみなされるとき。 |
| (2) | 催奇形性試験と対比して、繁殖試験の長期投与による雌動物への影響(薬物代謝酵素の誘導、肝毒性等)が大きく変化していないとみなされるとき。 |
|
| 注2) | 極めて稀な場合を除いて、げっ歯類及びウサギについては、母体数が16〜20匹の評価で、ある程度の整合性が試験間で得られている。母体数が16匹以下になると試験間の一貫性を欠き、20〜24匹以上でも整合性及び精度が大きく向上することはない。 |
|
| [6] | 発がん性試験
本試験は、げっ歯類に対し被験物質を経口投与した時に発がん性を示すかどうかの情報を得ることを目的とする。 |
| 1. | 動物種及び性
げっ歯類2種以上(通常、ラット、マウス又はハムスターが用いられる。)ついて実施する。雌雄の動物を原則として同数用いる。離乳後、馴化期間を経てできるだけ早い時期の同一週齢の動物、通常5〜6週齢の動物を用いる。
また、種及び系統の選択に当たっては、感染性疾患に対する抵抗性、寿命、既知発がん性物質に対する感受性等の特性が知られていて、一般的に実験動物として広く使用されているものとする。特に、自然発生腫瘍の発生頻度に関するデータが蓄積されている系統を選択する。
|
| 2. | 動物数
試験に用いる動物数は、1群当たり雌雄各50匹以上とする。各群への動物の割り付けには、体重層別等による適切な無作為抽出法を用いる。
ラットでは投与開始後24ヵ月、マウス及びハムスターでは投与開始後18ヵ月の時点で、腫瘍以外の原因による死亡率が50%以内であることが望ましい。
また、いずれの群においても、動物の10%以上が共食い又は飼育上の問題で失われてはならない。
なお、中間屠殺を予定する場合は、そのために必要な数を追加すること。
|
| 3. | 投与経路
被験物質の投与経路は、経口投与とし、通常、混餌投与又は飲水投与により行う。ただし、被験物質が飼料(飲水)中で不安定である場合、飼料(飲水)中の被験物質の分析が困難である場合、又は飼料(飲水)が忌避される場合等混餌(飲水)投与が困難な場合には強制投与を行うことも差し支えない。また、一定の条件下において、被験物質をマイクロカプセルに封入して投与しても差し支えない。
|
| 4. | 投与期間及び試験期間
被験物質の投与は、原則として週7日とし、ラットでは24ヵ月以上30ヵ月以内、マウス及びハムスターでは18ヵ月以上24ヵ月以内とする。
試験期間は、投与終了時又は投与終了後1〜3ヵ月までとするが、試験の最長期間は、ラットでは30ヵ月、マウス及びハムスターでは24ヵ月とし、最低用量群又は対照群の動物の性別の一方において累積死亡率が75%になった場合には、その時点でその性の生存例を屠殺し、その性について試験を終了する。
|
| 5. | 用量段階及び対照群
| (1) | 用量段階
対照群の他に少なくとも3段階の投与群を設ける。
各用量段階は、以下により、用量反応関係がみられるように設定する。なお、用量設定にあたっては、そのための90日間反復投与毒性試験の結果等を参考とすること。また、用量設定の根拠を示すこと。
| (1) | 最高用量
理想的には、最高用量は、腫瘍以外の原因で対照群に比して有意に死亡率が増加せず、毒性影響が認められる用量とする。
なお、混餌投与の場合は、栄養障害が起こらないようにすべきである。通常、飼料添加濃度5%(W/W)を超える投与量で実施する必要はない。また、強制投与の場合には、通常、技術的に投与できる最大量又は1000mg/kgで何ら毒性影響が認められない場合は、それ以上の投与量で実施する必要はない。
|
| (2) | 最低用量
原則として、最高用量の10%以上で設定されている。
しかしながら、食品への添加量等を勘案して設定することが望ましく、設定した最低用量が被験物質の想定されるヒトの摂取量からみて著しくかけ離れている場合には、最高用量の10%未満の用量を設けてもよい。
|
| (3) | 中間用量
中間用量は、最高用量と最低用量との等比中項をとることが望ましく、通常、群間の公比は2又は3とする。 |
|
| (2) | 対照群
対照群としては、被験物質の投与を行わないこと以外、すべての点で被験物質投与群と同一条件の群を設ける。
被験物質の投与に媒体等を使用する場合には、投与媒体量の最も多い用量群と同量の投与を行うこと。毒性に関する情報が十分に得られていない媒体等を使用する場合には、さらに無処置対照群を加える。 |
|
| 6. | 観察及び検査
次の(1)〜(4)の項目について実施する。
| (1) | 一般状態、体重、摂餌(摂水)量
全試験動物について、一般状態を毎日観察し、定期的に体重及び摂餌量(飲水投与の場合は摂水量)を測定する。
なお、体重及び摂餌(摂水)量の測定の頻度は、通常、次のとおりである。
| 体重: | 投与開始前及び投与開始後3カ月までは少なくとも週1回、その後は少なくとも4週に1回。 |
| 摂餌(飲水)量: | 投与開始前及び投与開始後3カ月までは少なくとも週1回、その後は少なくとも4週に1回。 |
なお、この測定はケージ毎に行ってもよい。
|
| (2) | 血液検査
投与終了後の剖検前に全生存動物につき採血して血液検査(赤血球数、白血球数等)を行う。屠殺時に血液塗抹標本を作製しておき、貧血、並びにリンパ節、肝臓、脾臓の腫大等、造血器腫瘍を予想させる例については塗抹標本を検索する。
|
| (3) | 剖検及び病理組織学的検査
| (1) | 試験期間中の死亡例は速やかに剖検し、器官・組織の肉眼的観察及び病理組織学的検査を行う。
なお、腫瘍性病変の記載に関しては、腫瘍発生に至る各種変化(前癌病変)の所見も付け加える必要がある(以下(2)及び(3)において同様)。病理組織学的検査は、通常、以下の器官の組織について行われるが、肉眼的観察所見等からの判断によって適宜追加する。
皮膚、乳腺、リンパ節(頚部リンパ節、腸間膜リンパ節等)、大動脈、唾液腺、骨及び骨髄(胸骨、大腿骨)、胸腺、気管、肺及び気管支、心臓、甲状腺及び上皮小体、舌、食道、胃、十二指腸、小腸(空腸、 回腸)、大腸(盲腸、結腸、直腸)、肝臓及び胆嚢、膵臓、脾臓、腎 臓、副腎、膀胱、精嚢、前立腺、精巣・精巣上体、卵巣及び卵管、子宮、膣、脳、下垂体、坐骨神経、骨格筋、脊髄、鼻腔(鼻甲介)、眼球及びその附属器、ジンバル腺、そのほか肉眼的に変化が認められた器官・組織
|
| (2) | 試験期間中に死に瀕した例は、速やかに隔離又は屠殺、剖検し、上記(1)と同様の観察、検査を行う。 |
| (3) | 試験終了時のすべての生存例は、速やかに屠殺、剖検し、器官・組織の肉眼的観察を行う。最高用量群及び対照群の全試験動物について上記(1)と同様に病理組織学的検査を行う。ただし、最高用量群と対照群の間で腫瘍発生率に差のある器官・組織が認められた場合には、他の用量群の全試験動物についても当該器官・組織の病理組織学的検査を行う。
なお、上述の場合のほかにあっても、全試験動物について病理組織学的検査を行うことは、評価の助けになる。 |
|
|
| [7] | 1年間反復投与毒性/発がん性併合試験
本試験は、長期間にわたり、被験物質を反復投与したときに発現する有害作用を検出するために行われるものであり、被験物質の1年間反復投与毒性と同時に発がん性に関する情報を得ることを目的とする。 |
| 1. | 動物種及び性
げっ歯類1種(通常、ラットが用いられる。)について実施する。雌雄の動物を原則として同数用いる。離乳後、馴化期間を経てできるだけ早い時期の同一週齢の動物、通常5〜6週齢の動物を用いる。
また、種及び系統の選択に当たっては、感染性疾患に対する抵抗性、寿命、既知がん原性物質に対する感受性等の特性が知られていて、一般的に実験動物として広く使用されているものとする。特に、自然発生腫瘍の発生頻度に関するデータが蓄積されている系統を選択する。
|
| 2. | 動物数
| A. | 発がん性検索のための試験に用いる動物数は、1群当たり雌雄各50匹以上とする。各群への動物の割り付けには、体重層別等による適切な無作為抽出法を用いる。ラットでは投与開始後24ヵ月、マウスでは投与開始後18ヵ月の時点で、腫瘍以外の原因による死亡率が50%以内であることが望ましい。また、いずれの群においても、動物の10%以上が共食い又は飼育上の問題で失われてはならない。なお、中間屠殺を予定する場合は、そのために必要な数を追加すること。
|
| B. | 1年間反復投与毒性検索のためにサテライト群を設けるが、サテライト群の動物数は、1群当たり雌雄各10匹以上とする。ただし、最高用量群については、20匹以上とする。 |
|
| 3. | 投与経路
被験物質の投与経路は、経口投与とし、通常、混餌投与又は飲水投与により行う。ただし、被験物質が飼料(飲水)中で不安定である場合、飼料(飲水)中の被験物質の分析が困難である場合、又は飼料(飲水)が忌避される場合等混餌(飲水)投与が困難な場合には強制投与を行うことも差し支えない。また、一定の条件下において、被験物質をマイクロカプセルに封入して投与しても差し支えない。
|
| 4. | 投与期間及び試験期間
被験物質の投与は、原則として週7日とし、ラットでは24ヵ月以上30ヵ月以内、マウスでは18ヵ月以上24ヵ月以内とする。
試験期間は、投与終了時又は投与終了後1〜3ヵ月までとするが、試験の最長期間は、ラットでは30ヵ月、マウスでは24ヵ月とし、最低用量群又は対照群の動物の性別の一方において累積死亡率が75%になった場合には、その時点でその性の生存例を屠殺し、その性について試験を終了する。
1年間反復投与毒性検索のためのサテライト群及びその対照群については、1年間反復投与毒性試験の試験期間に準じて差し支えない。
|
| 5. | 用量段階及び対照群
| (1) | 用量段階
| A. | 被験物質の発がん性検索のための用量段階
対照群の他に少なくとも3段階の投与群を設け、さらに、これらに合わせて1年間反復投与毒性検索のためのサテライト群及びその対照群を設ける。各用量段階は、以下により、用量反応関係がみられるように設定する。なお、用量設定にあたっては、そのための90日間反復投与毒性試験の結果等を参考とすること。また、用量設定の根拠を示すこと。 |
| (1) | 最高用量
理想的には、最高用量は、腫瘍以外の原因で対照群に比して有意に死亡率が増加せず、毒性影響が認められる用量とする。
なお、混餌投与の場合は、栄養障害が起こらないようにすべきである。通常、飼料添加濃度5%(W/W)を超える投与量で実施する必要はない。また、強制投与の場合には、通常、技術的に投与できる最大量又は1000mg/kgで何ら毒性影響が認められない場合は、それ以上の投与量で実施する必要はない。 |
| (2) | 最低用量
原則として、最高用量の10%以上で設定されている。
しかしながら、食品への添加量等を勘案して設定することが望ましく、設定した最低用量が被験物質の想定されるヒトの摂取量からみて著しくかけ離れている場合には、最高用量の10%未満の用量を設けてもよい。 |
| (3) | 中間用量
中間用量は、最高用量と最低用量との等比中項をとることが望ましく、通常、群間の公比は2又は3とする。 |
| B. | 被験物質の1年間反復投与毒性検索のための用量段階
対照群の他に少なくとも3段階の投与群を設ける。
用量段階は被験物質の毒性の全容を明らかにし、無毒性量(NOAEL)を推定することができるように設定する。したがって、最高用量は多数例の死亡を起こすことなく毒性影響が認められる用量、最低用量は何ら毒性影響が認められない用量とし、かつ用量反応関係がみられるように各用量段階を設定する。 |
|
| (2) | 対照群
対照群としては、被験物質の投与を行わないこと以外、すべての点で被験物質投与群と同一条件の群を設ける。
被験物質の投与に媒体等を使用する場合には、投与媒体量の最も多い用量群と同量の投与を行うこと。毒性に関する情報が十分に得られていない媒体等を使用する場合には、さらに無処置対照群を加える。 |
|
| 6. | 観察及び検査
次の(1)〜(6)の項目について実施する。
| (1) | 一般状態、体重、摂餌(摂水)量
全試験動物について、一般状態を毎日観察し、定期的に体重及び摂餌量(飲水投与の場合は摂水量)を測定する。
なお、体重及び摂餌(摂水)量の測定の頻度は、通常、次のとおりである。
| 体重: | 投与開始前及び投与開始後3カ月までは少なくとも週1回、その後は少なくとも4週に1回。 |
| 摂餌(摂水)量: | 投与開始前及び投与開始後3カ月までは少なくとも週1回、その後は少なくとも4週に1回。 |
なお、この測定はケージ毎に行ってもよい。
|
| (2) | 血液検査
発がん性検索のための用量群では、投与終了後の剖検前に全生存動物につき採血して血液検査(赤血球数、白血球数等)を行う。屠殺時に血液塗抹標本を作製しておき、貧血、並びにリンパ節、肝臓、脾臓の腫大等、造血器疾患を予想させる例については塗抹標本を検索する。
また、1年間反復投与毒性検索のためのサテライト群では全試験動物に対して、剖検前(12カ月時)に採血して血液学的検査及び血液生化学的検査を実施する。サテライト群についての検査は、通常、次の項目について検査される。なお、検査項目及び検査方法は、国際的に広く採用されているものを考慮に入れて選定するべきである。
血液学的検査:赤血球数、白血球数、血液像(白血球型別百分率)、血小板数、血色素、ヘマトクリット、その他必要に応じて網状赤血球、凝固能(プロトロンビン時間、活性化部分トロンボプラスチン時間)等血液生化学的検査:血清(血漿)総蛋白、アルブミン、A/G比、蛋白分画、ブドウ糖、コレステロール、トリグリセリド、ビリルビン、尿素窒素、クレアチニン、トランスアミナーゼ[AST(GOT),ALT(GPT)]、γ−GTP、アルカリホスファターゼ、電解質(ナトリウム、カリウム、塩素、カルシウム、無機リン等)等
|
| (3) | 尿検査
1年間反復投与毒性検索のためのサテライト群の全試験動物について、血液検査と同時期に尿検査を行う。
検査は、通常、次の項目について実施される。
尿量、pH、蛋白、糖、ケトン体、ビリルビン、ウロビリノーゲン、潜血、沈渣、比重又は浸透圧、電解質(ナトリウム、カリウム等)
|
| (4) | 眼科学的検査
1年間反復投与毒性検索のためのサテライト群の全試験動物について、必要に応じて、投与期間中少なくとも1回(12カ月時)、眼科学的検査を行う。
検査は、通常、肉眼的及び検眼鏡的に行い、前眼房・中間透光体・眼底のそれぞれについて実施される。
|
| (5) | その他の機能検査
必要に応じて、心電図、視覚、聴覚、腎機能等の検査を行う。
|
| (6) | 剖検及び病理組織学的検査
| (1) | 試験期間中の死亡例は速やかに剖検し、器官・組織の肉眼的観察及び病理組織学的検査を行う。
なお、腫瘍性病変の記載に関しては、腫瘍発生に至る各種変化(前癌病変)の所見も付け加える必要がある(以下(2)及び(3)において同様)。病理組織学的検査は、通常、以下の器官の組織について行われるが、肉眼的観察所見等からの判断によって適宜追加する。
皮膚、乳腺、リンパ節(頚部リンパ節・腸間膜リンパ節等)、大動脈、唾液腺、骨及び骨髄(胸骨、大腿骨)、胸腺、気管、肺及び気管支、心臓、甲状腺及び上皮小体、舌、食道、胃、十二指腸、小腸(回腸、空腸)、大腸(結腸、盲腸、直腸)、肝臓及び胆嚢、膵臓、脾臓、腎臓、副腎、膀胱、精嚢、前立腺、精巣・精巣上体、卵巣及び卵管、子宮、膣、脳、下垂体、坐骨神経、骨格筋、脊髄、鼻腔(鼻甲介)、眼球及びその附属器、ジンバル腺、そのほか肉眼的に変化が認められた器官・組織
|
| (2) | 試験期間中に死に瀕した例は、速やかに隔離又は屠殺、剖検し、上記(1)と同様の観察、検査を行う。 |
| (3) | 発がん性検索のための試験群及び1年間反復投与毒性検索のためのサテライト群に対しては、試験終了時にすべての生存例を、速やかに屠殺、剖検し、器官・組織の肉眼的観察を行う。なお、サテライト群では器官について重量測定を行う。最高用量群及び対照群の全試験動物について上記(1)と同様に病理組織学的検査を行う。ただし、最高用量群と対照群の間で腫瘍発生率に差のある器官・組織が認められた場合には、他の用量群の全試験動物についても当該器官・組織の病理組織学的検査を行う。
なお、上述の場合のほかにあっても、全試験動物について病理組織学的検査を行うことは、評価の助けになる。 |
|
|
| [8] | 抗原性試験
化学物質によるアレルギーは時として人体に重篤な障害を惹起することがあり、食品添加物についても、その安全性を確保するために抗原性(アレルギー原性)を検討する必要がある。
抗原性試験は、通常、次のような手法が用いられているが、化学物質を経口的に摂取した場合のアレルギー誘発能を予測する方法は十分に確立されていない。当面は被験物質の性質、使用形態等を考慮した上で、実験者が適切と判断した感作及び惹起方法で試験を実施すること。 |
| 1. | 即時型アレルギー試験
| (1) | モルモットにおける能動全身性アナフィラキシー反応試験 |
| (2) | ウサギ又はモルモットにおける同種PCA反応試験 |
| (3) | 感作マウス血清におけるラットPCA反応試験 |
|
| 2. | 遅延型アレルギー試験
| (1) | モルモットにおける接触皮膚反応試験 |
| (2) | マウスにおける足蹠反応又はリンパ節反応試験 |
なお、高分子又は蛋白質と結合すると考えられる食品添加物では、更に次の点を必要に応じて検討する。
| (1) | 感作動物血清の抗体力価 |
| (2) | 蛋白質との結合性の程度 |
| (3) | 類縁化合物との交差反応性 |
| (4) | その他 |
また、類似の化学物質で抗原性及びこれに起因すると考えられる作用が既に知られている場合には、それらに用いられた試験方法と同様な方法での検討もなされることが望ましい。
なお、報告に際しては、試験方法(使用動物、惹起抗原、対照群等)を明記し、かつ成績についての考察を行なうこと。 |
| [9] | 変異原性試験
本試験は、被験物質がDNAに影響を与え、その結果、遺伝子突然変異あるいは染色体の構造異常及び数的異常を起こす性質があるかどうかを明らかにすることを目的とする。
変異原性試験としては、「1.微生物を用いる復帰変異試験」、「2.哺乳類培養細胞を用いる染色体異常試験」、及び「3.げっ歯類を用いる小核試験」を実施する。なお、上記の試験結果を補足する必要がある場合には、適切な変異原性試験を追加して実施する。以下に追加試験を例示する。 |
| (1) | 遺伝子突然変異を指標とする試験
| (1) | 哺乳類培養細胞を用いる遺伝子突然変異試験 |
| (2) | ショウジョウバエを用いる遺伝子突然変異試験 |
| (3) | げっ歯類を用いる遺伝子突然変異試験 |
|
| (2) | 染色体異常を指標とする試験
| (1) | げっ歯類の骨髄細胞を用いる染色体異常試験 |
| (2) | げっ歯類の生殖細胞を用いる染色体異常試験 |
| (3) | げっ歯類を用いる優性致死試験 |
|
| (3) | DNA損傷を指標とする試験
| (1) | 微生物を用いるDNA修復試験 |
| (2) | 哺乳類の細胞を用いる不定期DNA合成(UDS)試験 |
| (3) | 哺乳類の細胞を用いる姉妹染色分体交換(SCE)試験 |
|
| 1. | 微生物を用いる復帰突然変異試験
微生物を用いる試験が不適切と考えられる場合(抗菌性の強い物質や哺乳類細胞に特異的に作用する物質等)には、微生物を用いる試験の代わりに、哺乳類培養細胞を用いる遺伝子突然変異試験を行うことが望ましい。
| (1) | 菌株
ネズミチフス菌(Salmonella typhimurium)の TA98,TA100,TA1535,{TA1537 あるいは TA97 あるいはTA97a}及び{大腸菌(Escherichia coli)の WP2 uvrA あるいはWP2 uvrA pKM101あるいはネズミチフス菌TA102}の5菌株を用いる。
|
| (2) | 用量段階
5段階以上の試験用量群を設定する。
|
| (3) | 最高用量
予備試験を行って抗菌作用の有無を求め、被験物質の溶解性にかかわらず、明らかな抗菌性を示す用量を最高用量とする。
抗菌作用あるいは変異原性が認められない場合は、5mg/プレート又は最低析出用量を限度とする。
|
| (4) | 対照
陰性対照は溶媒対照とする。陽性対照としては、既知変異原物質(S9mix)を必要としない物質と必要とする物質)を用いる。
|
| (5) | 代謝活性化
S9mixを加えた試験を並行して行う。哺乳類(通常はラット)に適切な薬物代謝酵素系の誘導剤を投与した後、肝臓からS9を調製する。このS9に補酵素等を加えたS9mixを用いる。
|
| (6) | 試験方法
プレインキュベーション法又はプレート法とする。
用量当り2〜3枚のプレートを用いる。
|
| (7) | 結果
復帰変異コロニー数の実測値とその平均値を表示する。 |
|
| 2. | 哺乳類培養細胞を用いる染色体異常試験
| (1) | 細胞
哺乳類の初代又は樹立培養細胞株を用いる。チャイニ−ズ・ハムスタ−培養細胞株(CHL、CHO等)やヒト培養リンパ球等が汎用されている。
|
| (2) | 用量段階
3段階以上の試験用量群を設定する。
|
| (3) | 最高用量
予備試験を行って細胞毒性の現れる用量を求める。被験物質の溶解性にかかわらず、単層培養細胞株では細胞増殖が50%以上抑制される用量、ヒト培養リンパ球では分裂頻度が50%以上抑制される用量を最高用量とする。
細胞毒性が認められない場合は、10mM、5mg/ml又は最低析出用量のいずれか低い濃度を限度とする。
|
| (4) | 対照
陰性対照は溶媒対照とする。陽性対照としては、既知染色体異常誘発物質(S9mixを必要としない物質と必要とする物質)を用いる。
|
| (5) | 代謝活性化
S9mixを加えた試験を並行して行う。哺乳類(通常はラット)に適切な薬物代謝酵素系の誘導剤を投与した後、肝臓からS9を調製する。このS9に補酵素等を加えたS9mixを用いる。
|
| (6) | 試験方法
S9mix添加及び非添加の条件で、それぞれ被験物質を短時間(例えば6時間)処理し、適切な時期(およそ正常細胞周期の1.5倍)に染色体標本を作製する。結果が共に陰性の場合にはS9mix非添加で長時間の連続処理(正常細胞周期の1.5倍あるいは3倍)後、直ちに標本を作製する。
用量あたり1〜2枚のプレートを用いる。用量当たり200個の分裂中期像について、染色体の構造異常の出現頻度を求める。構造異常は染色分体異常と染色体異常に分けて、その種類を明記する。
数的異常(倍数体や核内倍加等)が観察された場合には、その出現頻度を求める。
|
| (7) | 結果
構造異常を持つ細胞の出現頻度あるいは細胞当りの構造異常頻度、並びに数的異常を持つ細胞の出現頻度を表示する。 |
|
| 3. | げっ歯類を用いる小核試験
本試験をげっ歯類の骨髄細胞を用いる染色体異常試験で代行してもよい。
| (1) | 動物
マウス又はラットを用いる。毒性に明かな性差がみられない場合には、一方のみの性で十分である。その場合には雄の使用が望ましい。
|
| (2) | 動物数
1群5匹以上とする。
|
| (3) | 投与経路
強制経口投与又は腹腔内投与とする。
|
| (4) | 用量段階
3段階以上の試験用量群を設定する。
|
| (5) | 最高用量
最高用量は、幼若赤血球の減少等骨髄で何らかの細胞毒性の徴候が認められる用量、あるいは同じ投与プロトコールを用いて、それ以上の用量では致死が予想される用量とする。必要があれば予備試験を行う。毒性徴候が認められない場合は、2g/kgを限度とする。
|
| (6) | 対照群
陰性対照は溶媒対照とする。陽性対照としては、既知小核誘発物質を用いる。
|
| (7) | 投与回数
単回又は複数回投与とする。
|
| (8) | 試験方法
骨髄又は末梢血の赤血球を用いる。単回投与の場合は被験物質投与後適切な時期に2回、複数回投与の場合は最終投与後適切な時期に1回、標本を作製する。必要があれば予備試験を行い、最も感受性の高い時期を選択する。
アクリジン・オレンジ蛍光染色法又はギムザ染色法を用いる。
個体当り2000個の幼若赤血球について、小核を有する細胞の出現頻度を求める。同時に全赤血球に対する幼若赤血球の割合(%)を求める。
|
| (9) | 結果
小核を有する幼若赤血球の出現頻度及び全赤血球に対する幼若赤血球の割合(%)を個体別及び群別に表示する。 |
|
| [10] | 一般薬理試験
本試験は、被験物質の生体の機能に及ぼす影響を、主に薬理学的手法を用いて明らかにすることを目的とする。
本試験では、実施すべき試験項目を、A.原則として全ての被験物質について行うものと、B.前記Aの結果より判断し、必要に応じて行うものに分類した。
また、被験物質の化学構造や得られている毒性等の情報・知見から判断し、必要と思われる試験を追加実施することも考慮する。
なお、試験のあり方は、被験物質の特性によって異なることが十分考えられるので、試験項目及び試験方法は被験物質ごとに適切なものを選択する。 |
| 1. | 試験動物及び試験系
マウス、ラット、モルモット、ウサギ、ネコ、イヌ等各試験に適した動物種を用いる。なお、系統、性別、年齢等を考慮に入れる。
試験系としては、以下のものがある。
| (1) | 丸ごとの動物 |
| (2) | 摘出器官及び組織 |
| (3) | 血液及び血液成分 |
| (4) | 細胞及び細胞下レベル等 |
試験動物及び試験系の選択に当たっては、感受性、再現性、繁用性に留意する。
また、ヒトに対する予測性を考慮し、適切な情報が得られるような試験動物及び試験系を用いることが望ましい。
|
| 2. | 適用法
| (1) | 適用経路
丸ごと動物を用いる試験では、原則として、経口適用又はそれに準ずる経路とする。
ただし、試験の種類によっては被験物質の生体機能に及ぼす影響を検知するのに適した経路を用いる。例えば、摘出器官では栄養液への直接添加等がある。
なお、吸収の良くない被験物質の場合は、重要な試験項目については、静脈内適用等の適切な経路でも試験を行うことが望ましい。
|
| (2) | 適用回数
丸ごとの動物を用いる試験では、単回適用を原則とするが、反復適用による影響が予想される場合には、適切な回数の適用を行う。
|
| (3) | 用量設定
用量設定に当たっては、以下の点を考慮する。
| (1) | 用量作用関係を求め得る用量段階を設定する。 |
| (2) | 他の毒性試験において有害反応等を示す量から見て十分な量を用いる。
なお、体内動態試験等の情報・成績を考慮に入れ、選定した用量の妥当性を作用濃度、血中濃度等と関連づけて十分考察しておくことが望ましい。 |
|
| (4) | 対照群
対照群には、陰性対照(溶媒)群及び陽性対照(標準物質、作用既知の類似物質)群を適宜設ける。 |
|
| 3. | 試験項目
| A. | 原則としてすべての被験物質について行う項目で、生体機能に及ぼす影響の全体的な把握を目指した試験 |
| (1) | 一般症状及び行動に及ぼす影響
一般症状について観察する。
詳細な症状観察を行い、被験物質の作用を十分把握することに努める。
|
| (2) | 中枢神経に及ぼす影響
| (1) | 自発運動量に及ぼす影響を検討する。 |
| (2) | 麻酔作用について検討する。
無処置動物について被験物質の作用を検討するとともに、必要に応じて、バルビツール酸誘導体等による誘発処置との協力及び拮抗作用についても調べる。 |
| (3) | 痙攣作用について検討する。
無処置動物について被験物質の作用を検討するとともに、必要に応じて、電撃、ペンテトラゾール等による誘発処置との協力及び拮抗作用についても調べる。 |
| (4) | 痛覚に及ぼす影響を検討する。 |
| (5) | 体温に及ぼす影響を検討する。 |
|
| (3) | 自律神経系及び平滑筋に及ぼす影響
摘出回腸を用いて検討する。
被験物質単独の作用及びアゴニスト(ヒスタミン、アセチルコリン、塩化バリウム、セロトニン等)との相互作用を調べる。
|
| (4) | 呼吸・循環器系の及ぼす影響
呼吸運動、血圧、血流量、心拍数及び心電図に及ぼす影響を検討する。
通常は麻酔動物が用いられる。必要に応じ、無麻酔動物についても調べる。
|
| (5) | 消化器系に及ぼす影響
胃腸管内輸送能に及ぼす影響を検討する。
腸管内輸送能について調べるが、必要に応じて胃内容排出能についても検討する。
|
| (6) | 水及び電解質代謝に及ぼす影響
尿量、尿中ナトリウム・カリウム・塩素イオン濃度を測定する。
|
| (7) | その他の重要な作用
類似の化学構造又は作用を有する既知物質の作用から予想される作用で、A.の(1)〜(6)で検討されないもの。 |
| B. | Aの試験結果より判断して、必要に応じて行う試験 |
| (1) | 中枢神経系に及ぼす影響
| (1) | 自発脳波に及ぼす影響を検討する。
必要に応じ、得られたデータについて機器を用いて解析を行う。 |
| (2) | 脊髄反射に及ぼす影響を検討する。 |
| (3) | 条件回避反応に及ぼす影響を検討する。 |
| (4) | 協調運動に及ぼす影響を検討する。 |
|
| (2) | 体性神経系に及ぼす影響
| (1) | 神経・筋接合部に及ぼす影響を検討する。 |
| (2) | 筋弛緩作用について検討する。 |
| (3) | 局所麻酔作用について検討する。 |
|
| (3) | 自律神経系及び平滑筋に及ぼす影響
| (1) | 瞳孔径及び瞬膜収縮に及ぼす影響を検討する。 |
| (2) | 血管、器官、輸精管、子宮等の摘出器官・組織を用いて検討する。 |
|
| (4) | 呼吸・循環器系に及ぼす影響
| (1) | 自律神経作用薬並びに迷走神経刺激及び総頚動脈閉塞等による血圧及び心拍数の変化に対する作用を検討する。 |
| (2) | 生体位心臓を用いて検討する。 |
| (3) | 心臓、心房、乳頭筋、血管床等の摘出器官・組織を用いて検討する。 |
|
| (5) | 消化器系に及ぼす影響
| (1) | 胃液、唾液、胆汁及び膵液分泌に及ぼす影響を検討する。 |
| (2) | 摘出胃・腸管を用い、その運動に及ぼす影響を検討する。 |
| (3) | 生体位胃・腸管を用い、その運動に及ぼす影響を検討する。 |
| (4) | 胃・十二指腸粘膜に対する作用を検討する。 |
|
| (6) | その他の作用
| (1) | 血液凝固系に及ぼす影響を検討する。 |
| (2) | 血小板凝集に対する作用を検討する。 |
| (3) | 溶血作用について検討する。 |
| (4) | 腎機能に及ぼす影響を検討する。 |
|
|
| [11] | 体内動態試験
本試験は、被験物質を動物に投与してその吸収、分布、代謝及び排泄等体内動態に関する情報を得ることを目的とする。本試験の資料は毒性試験あるいはその結果の評価に資する。
本試験は以下に述べる原則を参考にして行う。ただし、被験物質の性質に応じて適切な方法を考慮し、試験の目的に沿うように、適宜取捨選択、又は他の方法に置き換えても差し支えない。また、被験物質の体内動態に関する適切なデータが毒性試験から得られた場合には、これを利用しても良い。 |
| 1. | 被験物質
食品添加物、食品添加物として開発を意図する化合物又はそれらの同位元素 標識体を使用する。なお、同位元素標識体にあっては、入手先、合成法、純度、標識核種、標識位置、比放射能、安定性等を明確にしておく。
|
| 2. | 動物種、性
げっ歯類1種以上(通常、ラットが用いられる。)及び非げっ歯類1種以上(通常、イヌが用いられる。)の合計2種以上について実施することが望ましい。毒性試験との対応を考えて、適切な動物を選定する。
なお、毒性試験に用いたものと同一の系統であることが望ましい。また、原則として雌雄の動物について検討する。必要に応じて、幼若動物についても検討する。
|
| 3. | 動物数
動物数は、個体差、各観察・測定時点での必要検体数等を考慮して試験目的にあった適切な数とする。
|
| 4. | 投与経路
原則として経口投与とする。なお、必要に応じて、静脈内投与等による試験を補足する。
|
| 5. | 投与回数
単回投与及び反復投与を行う。
反復投与の場合には、動物試験における被験物質の体内での定常状態、蓄積性等を推定し得るに足る投与間隔と期間で投与する。
なお、反復投与については、他の毒性試験によって得られる体内動態に関する情報を利用してもよい。
|
| 6. | 用量段階
2段階以上とする。
2段階の用量設定に当たっては、反復投与毒性試験の最高用量及び何ら毒性影響が認められない用量(無毒性量;NOAEL)を目安とする。
なお、低用量段階の設定に当たっては、可能ならば食物経由により摂取することが推定される量を考慮する。
|
| 7. | 定量法
定量の方法及びその感度、精度、特異性等を明確にする。
|
| 8. | 検討項目
吸収、分布、代謝、排泄の各段階における検討項目は、通常、次のとおりである。なお、生物学的半減期(もしくはこれに準ずる定数)、クリアランス、分布容積及び生物学的利用能等の体内動態に関するパラメータを必要に応じて求めるとともに、体内動態の非線形性の有無を検討する。また、生体内で代謝等を受ける場合においては、代謝物についても検討する。
| (1) | 吸収
被験物質の吸収量及び吸収速度に関する情報を求める。これらは、血中濃度−時間曲線又は累積排泄量曲線等の解析から求められる。 |
| A. | 血中濃度(血清中濃度、血漿中濃度又は全血中濃度)−時間曲線による方法
吸収の程度と速度は、投与後の最高血中濃度(Cmax)、そのときの時間(Tmax)、血中濃度−時間曲線下面積(AUC)等を解析することにより求めることができる。
また、これらのパラメータと、静脈内投与又はその他基準となる投与方法により得た同様のパラメータとを比較することにより、吸収の程度と速度をより明確に推定することができる。
|
| B. | 累積排泄量曲線による方法
尿、糞、胆汁、呼気等への排泄量を測定し、これらにより総排泄量を求める。これらは吸収量のよい尺度となることが多い。
また、必要に応じて、吸収に影響する次の要因も検討する。
| (1) | 食物中での存在形態 |
| (2) | 吸収部位 |
| (3) | 消化管内での代謝、安定性 |
| (4) | 食餌、消化管内pH |
|
| (2) | 分布
被験物質の各種器官及び組織への分布、その経時的変化及び蓄積性に関する情報を求める。なお、体内動態を適切に反映する数時点での測定が望ましい。
| (1) | 器官内及び組織内濃度
反復投与により高濃度分布又は蓄積のみられた器官及び組織、有害反応に関わる器官及び組織については、その存在形態についても検討することが望ましい。
なお、全身オートラジオグラフィーは器官及び組織を摘出して測定する方法では明らかにし難い部位への被験物質の分布に関する情報を得るためには有効な手段である。 |
| (2) | 胎盤、胎児、乳汁への移行性 |
| (3) | 血漿中の蛋白との結合、血球への分配 |
|
| (3) | 代謝
被験物質及びその主要な代謝物の同定と定量を行い、代謝経路及び代謝の程度と速度に関する情報を求める。
本試験は、通常、血液、尿、胆汁及び糞等の生体試料中の未変化体と代謝物を分離定量することによって行われる。代謝に関与する器官のスライス、ホモジネート、細胞懸濁液、細胞分画等の試料を用いたin vitro 試験は、in vivo 試験とともに代謝試験として有用である。
|
| (4) | 排泄
被験物質及びその主要な代謝物の排泄経路及び排泄の程度と速度に関する情報を求めるため、次の経路について検討する。
| (1) | 尿、糞、呼気
放射同位元素標識体を単回投与した場合には、投与後7日間又は投与した放射能の少なくとも95%が回収されるまで測定することが望ましい。 |
| (2) | 胆汁
主要排泄経路が胆汁の場合には、腸肝循環についても検討する。 |
| (3) | 乳汁 |
| (4) | 必要に応じて、排泄に影響する次の要因も検討する。
|
|
| (5) | 一般的留意点
以上の結果を総合し、また、類似の化学物質の既報文献とも比較考察する等幅広く検討する。なお、被験物質がラセミ体である場合には、毒性との関連において必要があれば、それぞれの光学異性体についても体内動態を検討することが望ましい。 |
|
ホーム > 政策について > 分野別の政策一覧 > 健康・医療 > 食品 > 食品添加物 > 食品添加物の指定及び使用基準改正に関する指針について > 食品添加物の指定及び使用基準改正に関する指針(別添)